Anatomia del pancreas

Vincenzo Triggiani
Endocrinologia e Malattie Metaboliche, Dipartimento Interdisciplinare di Medicina, Università degli Studi di Bari “Aldo Moro”
Il pancreas è una voluminosa ghiandola, con duplice funzione esocrina ed endocrina, annessa all’apparato digerente, avente forma allungata in senso trasversale e appiattita in senso sagittale, situata in posizione mediana, all’altezza delle prime due vertebre lombari, nello spazio retro-peritoneale, in corrispondenza della regione epigastrica. Presenta una guaina connettivale, dalla quale originano sottili setti che suddividono il parenchima in lobuli. Misura 12-15 cm in lunghezza, 4 cm in altezza e 1.5-2 cm in spessore, mentre il peso è di 70-110 g.
È possibile suddividerlo in tre porzioni.
- La testa, la parte più voluminosa, accolta all’interno della “C” duodenale, che si prolunga inferiormente nel processo uncinato; è rivestita anteriormente dal peritoneo ed è in rapporto con la parte pilorica dello stomaco e la parte superiore del duodeno. Posteriormente è rivestita dalla fascia retro-pancreatica, che la separa dal coledoco, dalla vena porta e dalla vena cava inferiore. La zona di passaggio tra testa e corpo è denominata istmo pancreatico.
- Il corpo, leggermente obliquo dal basso verso l'alto, contrae rapporto anteriormente con il peritoneo della borsa omentale e con la parete posteriore dello stomaco. Posteriormente è rivestito dalla fascia retro-pancreatica ed è in rapporto con la vena mesenterica superiore, l’aorta, la ghiandola surrenale e il rene sinistro.
- La coda si estende fino alla faccia gastrica della milza e posteriormente è in rapporto con il rene sinistro, ed è collegata all’ilo della milza dal legamento pancreatico-lienale.
Il pancreas è irrorato dalle arterie pancreatico-duodenali superiori e inferiori, che provengono dall’arteria celiaca, e da vasi che originano dall’arteria splenica. Il drenaggio venoso è tributario del sistema portale: le vene pancreatiche drenano corpo e coda e sboccano nella vena splenica; le vene pancreatico-duodenali sboccano nella vena splenica o direttamente nella vena porta. La rete linfatica, particolarmente ricca, drena nei linfonodi pancreatico-lienali e celiaci.
L’innervazione efferente è sotto il controllo vagale, mentre le vie afferenti decorrono lungo i nervi splancnici.
La componente esocrina rappresenta circa l’80% della ghiandola. Le subunità funzionali del pancreas esocrino sono gli acini, delimitati da cellule secernenti. Nel lume acinare si raccoglie il secreto, che poi fluisce nei dotti intra-lobulari e quindi nei dotti inter-lobulari, che sboccano a loro volta nel dotto pancreatico principale di Wirsung. Questo inizia in corrispondenza della coda e percorre tutta la ghiandola, aumentando di calibro, si unisce al coledoco, per poi sboccare a livello della seconda porzione del duodeno, in corrispondenza della papilla duodenale maggiore (di Vater), attraverso lo sfintere di Oddi. Dal dotto principale origina a livello dell’istmo il dotto pancreatico accessorio (di Santorini) che attraversa la testa del pancreas per sfociare nel duodeno in corrispondenza della papilla duodenale minore, circa 2 cm sopra l’ampolla di Vater.
Il succo pancreatico prodotto dalla componente esocrina è costituito da costituenti inorganici, quali acqua, sodio, potassio, cloro e bicarbonato, e costituenti organici, gli enzimi lipasi, amilasi, proteasi, ribonucleasi e desossiribonucleasi, deputati alla digestione dei nutrienti. Per proteggere il pancreas dall’autodigestione, gli enzimi proteolitici sono prodotti in forma inattiva, per poi essere attivati nel lume intestinale.
La componente endocrina rappresenta circa il 2% dell’organo ed è costituita dalle isole di Langerhans, collocate prevalentemente a livello del corpo-coda. Sono costituite da diversi tipi cellulari:
- le cellule α (20%) producono glucagone;
- le cellule β (75%) producono insulina;
- le cellule δ producono somatostatina;
- le cellule PP producono polipeptide pancreatico;
- le cellule ε producono ghrelina.
Bibliografia
- Stranding S. Anatomia del Gray. Le basi anatomiche per la pratica clinica, Ed. Elsevier, 2009.
Fisiologia del metabolismo glucidico
Vincenzo Triggiani
Endocrinologia e Malattie Metaboliche, Dipartimento Interdisciplinare di Medicina, Università degli Studi di Bari “Aldo Moro”
Il glucosio ha una posizione centrale nel metabolismo, sia perché è un eccellente combustibile, sia perché da esso deriva una varietà di composti metabolici intermedi per le biosintesi (figura). Esso può infatti essere:
- utilizzato per la sintesi di polisaccaridi complessi destinati allo spazio extra-cellulare;
- immagazzinato nella cellula sotto forma di glicogeno;
- ossidato a piruvato tramite la glicolisi per produrre ATP e intermedi metabolici;
- ossidato attraverso la via del pentosio-fosfato per produrre ribosio-5-fosfato necessario per la sintesi degli acidi nucleici e NADPH usato per i processi biosintetici riduttivi.
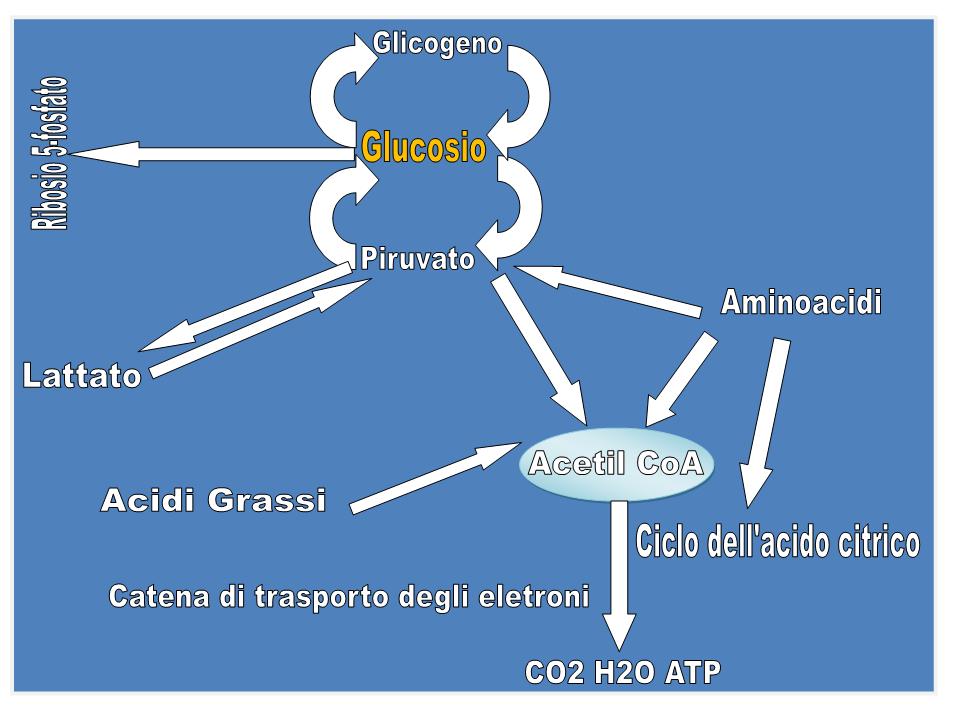
Nella glicolisi, una molecola di glucosio viene degradata, mediante una serie di reazioni citosoliche catalizzate da dieci enzimi, a 2 molecole di piruvato, mentre parte dell’energia rilasciata viene recuperata sotto forma di ATP e NADH. Per ogni molecola di glucosio trasformata in 2 molecole di piruvato la resa netta è di 2 molecole di ATP e 2 di NADH. In condizioni aerobiche, il piruvato viene ossidato ad acetato che entra sotto forma di acetil-CoA nel ciclo di Krebs (ciclo dell’acido citrico), per essere ossidato a CO2, grazie all'intervento del NAD+ e del FAD che si riducono a NADH e FADH2. La catena respiratoria, un complesso sistema ossido-riduttivo, nel processo della respirazione mitocondriale riossida i coenzimi ridotti, trasferendo all'ossigeno gli elettroni sottratti, con formazione di H2O e conservazione dell’energia rilasciata sotto forma di ATP (fosforilazione ossidativa). Quando invece i tessuti non possono essere riforniti di una quantità di O2 sufficiente per ossidare il piruvato e il NADH derivanti dalla glicolisi, come avviene in caso di un esercizio muscolare violento, il NAD+ viene rigenerato dal NADH per mezzo della riduzione del piruvato a lattato che è catalizzata dalla lattato-deidrogenasi. Il lattato viene poi riconvertito in glucosio dal fegato nella fase di recupero dopo l’attività muscolare intensa. Se il lattato è prodotto in grande quantità (ad es. in una corsa veloce), l’acidificazione conseguente alla ionizzazione dell’acido lattico nel muscolo e nel sangue limita la capacità di contrarsi del muscolo stesso. Gli eritrociti, non avendo mitocondri, anche in condizioni di aerobiosi non possono ossidare il piruvato a CO2 e quindi producono lattato. La glicolisi è la sola fonte di energia metabolica per alcuni tessuti come il cervello, la midollare renale, gli eritrociti e gli spermatozoi.
Il glucosio deriva soprattutto dall’amido contenuto nella dieta (cereali e legumi in particolare), mentre il glicogeno contenuto negli alimenti di origine animale (fegato e muscolo) contribuisce in minima parte. L’amido viene attaccato dalle amilasi salivare e soprattutto pancreatica e il glucosio liberato viene assorbito dall’epitelio intestinale e passa nel sangue, stimolando la secrezione pancreatica di insulina, che fa entrare il glucosio nel muscolo e nel tessuto adiposo e stimola sia l’ossidazione del glucosio che la sintesi di glicogeno, mentre inibisce la scissione del glicogeno a glucosio. L’ossidazione di un grammo di glucidi genera all’incirca 4 kcal.
Glucosio deriva anche da disaccaridi, come saccarosio e lattosio. Altri monosaccaridi (fruttosio, galattosio e mannosio) entrano nella glicolisi in punti diversi.
Oltre che da fonti esogene, il glucosio può derivare dalla degradazione del glicogeno endogeno, forma di deposito del glucosio all’interno soprattutto degli epatociti e del muscolo, derivante dall’azione della glicogeno-sintetasi, attivata dall’insulina secreta in risposta al pasto. Il glicogeno muscolare viene demolito dalla glicogeno-fosforilasi muscolare attivata dall’adrenalina, dagli ioni calcio e dal cAMP durante l’attività muscolare e il glucosio liberato viene utilizzato per le esigenze del muscolo stesso in condizioni di aerobiosi o anaerobiosi. Il glicogeno epatico viene degradato dalla glicogeno-fosforilasi, attivata dal glucagone, a glucosio che può passare in circolo per le esigenze dei tessuti, in particolare del sistema nervoso, tra un pasto e l’altro e nel digiuno.
Il fegato (e in minor misura il rene e le cellule dell’epitelio intestinale) può produrre glucosio, oltre che da lattato e piruvato, anche a partire da precursori non glucidici (gluconeogenesi), come il glicerolo e quegli aminoacidi (glucogenici) che possono formare piruvato o altri intermedi del ciclo di Krebs. Il glucosio può essere sintetizzato dai trigliceridi, più esattamente dal glicerolo derivante dalla scissione di questi, ma non dagli acidi grassi, in quanto il catabolismo di questi porta alla formazione di acetil-CoA, che non può essere trasformato in piruvato perché la reazione della piruvato-deidrogenasi è irreversibile. La gluconeogenesi condivide 7 delle 10 tappe della glicolisi, che avvengono in direzione opposta. Essa è energeticamente dispendiosa ma essenziale: la trasformazione di 2 molecole di piruvato in una di glucosio richiede infatti 4 molecole di ATP e 2 di GTP, ma in tal modo viene garantito un adeguato rifornimento di glucosio anche quando non c’è apporto con la dieta e sono state esaurite le riserve di glicogeno.
Una parte del glucosio viene ossidata nella via del pentosio-fosfato, che porta alla sintesi di ribosio-5-fosfato, zucchero fosforilato precursore della sintesi dei nucleotidi (sintesi di acidi nucleici, ATP, NADH, FADH2, CoA) e alla produzione di NADPH, riducente utilizzato per la sintesi di acidi grassi, colesterolo, ormoni steroidei, e per contrastare gli effetti dannosi dei radicali liberi dell’ossigeno.
Bibliografia
- Nelson DL, Cox MM. Introduzione alla biochimica di Lehninger. 4° edizione. Zanichelli Editore.
Fisiologia del metabolismo lipidico
Vincenzo Triggiani
Endocrinologia e Malattie Metaboliche, Dipartimento Interdisciplinare di Medicina, Università degli Studi di Bari “Aldo Moro”
I lipidi rappresentano la forma principale di conservazione dell’energia e sono i principali costituenti delle membrane cellulari.
Le cellule possono ottenere acidi grassi da ossidare da tre fonti: i grassi della dieta, i lipidi depositati nelle cellule in forma di gocce insolubili e i lipidi sintetizzati in un organo ed esportati in un altro.
I trigliceridi forniscono più di metà dell’energia consumata da alcuni organi, in particolare fegato, cuore e muscolo scheletrico a riposo. I trigliceridi della dieta, per poter essere assorbiti dall’epitelio intestinale, devono venire a contatto con i sali biliari con i quali formano micelle finemente disperse. In tal modo aumenta enormemente la frazione di lipidi accessibile all’azione delle lipasi (soprattutto la lipasi pancreatica), che convertono i trigliceridi in mono e digliceridi, acidi grassi e glicerolo. Questi prodotti vengono riconvertiti in trigliceridi nelle cellule intestinali e combinati con il colesterolo della dieta e con specifiche proteine a formare i chilomicroni. Queste lipoproteine passano attraverso il vaso chilifero contenuto nel microvillo intestinale nel sistema linfatico e quindi, attraverso il dotto toracico nella vena succlavia sinistra e quindi nel sangue, raggiungendo, senza passare dal fegato, i muscoli e il tessuto adiposo. Nei capillari di questi tessuti la lipoproteina-lipasi idrolizza i trigliceridi ad acidi grassi e glicerolo, che entrano nelle cellule. Nel muscolo gli acidi grassi vengono ossidati per produrre energia, nel tessuto adiposo vengono ri-esterificati a trigliceridi di deposito. Le rimanenze dei chilomicroni, contenenti colesterolo e apo-lipoproteine e scarsi trigliceridi, vengono internalizzate dagli epatociti, sui quali riconoscono specifici recettori per le apolipoproteine.
Adrenalina e glucagone, secreti in risposta a una bassa disponibilità di glucosio nel sangue, attivano l’adenilato-ciclasi a livello della membrana degli adipociti, determinando l’attivazione di protein-chinasi cAMP-dipendente, che da un lato attiva la perilipina A, che trasferisce la lipasi ormone-sensibile dal citosol alla superficie delle gocce lipidiche, dove inizia ad idrolizzare i trigliceridi in acidi grassi liberi e glicerolo, dall’altro attiva la lipasi stessa. Gli acidi grassi liberi escono dall’adipocita e si legano all’albumina, che li trasporta ai tessuti. Nelle cellule vengono ossidati, mentre il glicerolo viene convertito a di-idrossi-acetone fosfato ed entra nella via glicolitica.
L’ossidazione degli acidi grassi liberi avviene nei mitocondri. Quelli con catena fino a 12 atomi di carbonio entrano direttamente nel mitocondrio, mentre quelli a catena più lunga (la maggior parte di quelli che provengono dalla dieta o dal tessuto adiposo) ha bisogno del sistema shuttle della carnitina per attraversare la membrana mitocondriale. La ß-ossidazione è caratterizzata dalla rimozione ossidativa di unità bicarboniose sotto forma di acetil-CoA, iniziando dall’estremità carbossi-terminale. L’unità acetilica dell’acetil-CoA viene ossidata a CO2 nel ciclo di Krebs. L’energia rilasciata viene immagazzinata sotto forma di ATP nella fosforilazione ossidativa. L’ossidazione di un grammo di lipidi genera 9 kcal.
Nel fegato gli acetil-CoA sintetizzati a livello citosolico possono andare incontro alla ß-ossidazione nei mitocondri o essere convertiti a trigliceridi o fosfolipidi nel citosol. Quando l’organismo è ben rifornito di carboidrati, aumenta la concentrazione di malonil-CoA, che blocca il trasferimento di acidi grassi nel mitocondrio e, quindi, la loro ossidazione. L’eccesso di glucosio, che non può essere ossidato o convertito in glicogeno, viene trasformato in acidi grassi e quindi in trigliceridi sotto stimolo insulinico. L’acetil-CoA, formato nel fegato durante l’ossidazione degli acidi grassi, quando non può entrare nel ciclo dell’acido citrico (carenza di glucosio come nel digiuno) viene trasformato in corpi chetonici, che non possono essere metabolizzati dal fegato. Di questi, l’acetone viene eliminato con la respirazione, mentre l’aceto-acetato e il D-ß-idrossi-butirrato vengono trasportati dal sangue ai tessuti extra-epatici, come muscolo scheletrico, cuore, corticale renale, dove vengono ossidati nel ciclo dell’acido citrico. Il cervello, che normalmente ossida glucosio, in condizioni di digiuno può ossidare i corpi chetonici. Quando presenti in eccesso, i corpi chetonici danno luogo a una condizione di acidosi metabolica.
Per quanto riguarda la biosintesi degli acidi grassi, le lunghe catene carboniose vengono sintetizzate a partire dall’acetil-CoA prodotto nei mitocondri dall’ossidazione del piruvato o derivante dallo scheletro carbonioso degli aminoacidi (quello derivante dall’ossidazione degli acidi grassi invece non è disponibile, perché le due vie ossidativa e biosintetica sono regolate in modo coordinato e complementare), mediante la ripetizione di 4 tappe catalizzate nel citosol da un sistema multi-enzimatico chiamato acido grasso-sintetasi. Ad ogni passaggio, la catena si allunga di due atomi di carbonio. L’agente riducente è il NADPH generato nella via del pentosio-fosfato. Il principale prodotto è il palmitato, da cui derivano poi gli acidi grassi a catena lunga, mediante l’aggiunta successiva di unità acetili, catalizzata nel reticolo endoplasmatico liscio e nei mitocondri dal sistema di allungamento degli acidi grassi. Dal palmitato derivano anche i più comuni acidi grassi insaturi, mentre linoleato e alfa-linolenato, precursori di altri acidi grassi insaturi, devono essere introdotti con la dieta e, pertanto, vengono definiti acidi grassi essenziali.
Dagli acidi grassi poli-insaturi a 20 atomi di carbonio derivano gli eicosanoidi: prostaglandine, trombossani e leucotrieni.
Gli acidi grassi sintetizzati o ingeriti vengono incorporati nei trigliceridi, per la conservazione dell’energia metabolica, o nei fosfolipidi componenti delle membrane. L’insulina favorisce la conversione dei carboidrati in trigliceridi.
Biosintesi e degradazione dei trigliceridi sono regolate in modo coordinato e complementare: la via che viene favorita dipende dalle risorse metaboliche dell’organismo in un dato momento.
Il colesterolo è un componente essenziale delle membrane cellulari, precursore degli acidi biliari e degli ormoni steroidei. Deriva in parte dalla dieta, in parte dalla sintesi endogena a partire dall’acetato, dal quale derivano tutti i suoi atomi di carbonio. La maggior parte viene prodotta dal fegato. Una piccola parte è incorporata nelle membrane degli epatociti, mentre la quota maggiore viene esportata come colesterolo biliare, acidi biliari o esteri del colesterolo. Questi ultimi si formano per l’azione dell’acetil-CoA-colesterolo-aciltransferasi, sono idrofobici e sono conservati nel fegato o trasportati insieme ad altri lipidi sotto forma di lipoproteine dal fegato a quei tessuti che utilizzano il colesterolo.
Colesterolo, esteri del colesterolo, trigliceridi e fosfolipidi, pertanto, essendo insolubili in acqua, per essere trasportati ai tessuti che li utilizzano, devono essere aggregati a proteine trasportatrici, le apo-lipoproteine, a formare le lipoproteine. Diverse combinazioni di tipi diversi di apo-lipoproteine e lipidi danno origine a diverse classi di lipoproteine, con diversa densità e dimensioni.
I chilomicroni sono le lipoproteine più grandi e meno dense, contenendo elevate quantità di trigliceridi di derivazione alimentare, che trasportano ai tessuti dove vengono ossidati (muscolo) o depositati (tessuto adiposo). Dopo che i chilomicroni hanno perso i trigliceridi (chilomicroni remnant), arrivano al fegato, dove rilasciano colesterolo e vengono degradati.
L’eccesso di carboidrati della dieta può essere trasformato in trigliceridi dal fegato ed esportato, attraverso le lipoproteine a densità molto bassa (very low density lipoproteins o VLDL), contenenti oltre a trigliceridi anche colesterolo ed esteri di colesterolo, al muscolo e al tessuto adiposo, dove la lipoprotein-lipasi libera dai trigliceridi acidi grassi. Le VLDL divengono in tal modo lipoproteine a densità intermedia (intermediate density lipoproteins o IDL) che, con l’ulteriore rimozione di trigliceridi, divengono lipoproteine a bassa densità (low density lipoproteins o LDL), ricche in colesterolo ed esteri del colesterolo, che si legano a specifici recettori sulle cellule che utilizzano colesterolo (endocitosi mediata da recettore).
Le lipoproteine ad alta densità (high density lipoproteins o HDL) vengono sintetizzate dal fegato e dalle cellule intestinali come particelle piccole, ricche di proteine, con piccole quantità di colesterolo e prive di esteri del colesterolo, contenenti l’enzima lecitina-colesterolo-acil-transferasi, che catalizza la formazione di esteri del colesterolo utilizzando la lecitina (fosfatidil-colina). Queste particelle si caricano di colesterolo a livello dei tessuti periferici e riportano il colesterolo al fegato (trasporto inverso del colesterolo), dove in parte viene convertito in sali biliari.
L’insulina attiva, mentre il glucagone inibisce la sintesi del colesterolo. Entrambi agiscono sull’enzima che catalizza la tappa limitante la sintesi (HMG-CoA-reduttasi).
Subito dopo un pasto ricco di calorie, giungono al fegato glucosio, acidi grassi e aminoacidi. L’aumento della glicemia stimola la secrezione pancreatica di insulina, che a sua volta stimola l’assunzione di glucosio da parte del tessuto muscolare (glicolisi e sintesi di glicogeno) e del tessuto adiposo (glicolisi e sintesi di acidi grassi e trigliceridi). I grassi della dieta raggiungono invece il tessuto adiposo direttamente dall’intestino con i chilomicroni. Nel fegato l’insulina determina attivazione della glicogeno-sintetasi e inibizione della glicogeno-fosforilasi; attiva anche l’ossidazione del glucosio a piruvato, che viene ossidato ad acetil-CoA. Questo non viene ulteriormente ossidato, ma utilizzato per la sintesi degli acidi grassi, che in forma di trigliceridi (VLDL) vengono esportati nel tessuto adiposo e nei muscoli. Gli aminoacidi vengono utilizzati per la sintesi proteica nel fegato e negli altri tessuti, mentre gli aminoacidi in eccesso vengono convertiti in piruvato e in acetil-CoA usato per la sintesi lipidica. L’insulina stimola la sintesi e il deposito di trigliceridi nel tessuto adiposo.
A distanza di qualche ora dal pasto, i livelli di glucosio nel sangue tendono a ridursi, per la continua utilizzazione da parte del cervello e degli altri tessuti. Si riduce il rilascio di insulina e viene secreto glucagone, che determina aumento del rilascio di glucosio da parte del fegato attraverso l’attivazione della glicogeno-fosforilasi, che demolisce il glicogeno, l’inattivazione della glicogeno-sintetasi, l’inibizione della glicolisi epatica e la stimolazione della gluconeogenesi, che utilizza il piruvato, il lattato, il glicerolo derivante dai trigliceridi e gli aminoacidi glucogenici derivanti dal catabolismo proteico. A livello del tessuto adiposo stimola la lipasi ormono-sensibile, che libera dai trigliceridi di deposito glicerolo (utilizzato dalla gluconeogenesi) e acidi grassi, che vengono ossidati dal fegato e da altri tessuti, con risparmio di glucosio, che può restare disponibile per il cervello.
Quando il digiuno si protrae e l’apporto di glucosio al cervello inizia a ridursi, i corpi chetonici prodotti dal fegato divengono una fonte di energia fondamentale per vari tessuti, oltre che per il cervello stesso.
Anche l’adrenalina attiva la glicogeno-fosforilasi e inibisce la glicogeno-sintetasi, stimolando la liberazione di glucosio. Promuove anche l’utilizzazione anaerobica del glicogeno nel muscolo, fornendo energia di pronta utilizzazione. Determina anche la mobilizzazione di acidi grassi liberi dal tessuto adiposo. Stimola, inoltre, la secrezione di glucagone e inibisce la secrezione insulinica, favorendo la mobilizzazione di combustibili metabolici e inibendone il deposito. Ciò è importante, ad esempio, durante l’attività fisica e comunque quando si ha una riduzione della glicemia.
In varie situazioni di stress, il cortisolo stimola la scissione dei trigliceridi nel tessuto adiposo, determinando liberazione di acidi grassi, disponibili per l’ossidazione, e glicerolo, trasformato in glucosio dalla gluconeogenesi.
Bibliografia
- Nelson DL, Cox MM. Introduzione alla biochimica di Lehninger. 4° edizione. Zanichelli Editore.
Fisiologia dell'insulina e della contro-regolazione
Vincenzo Triggiani
Endocrinologia e Malattie Metaboliche, Dipartimento Interdisciplinare di Medicina, Università degli Studi di Bari “Aldo Moro”
L’organismo ha un continuo bisogno di energia per:
- l’attività di base (metabolismo basale) dei vari organi e tessuti (pompe ioniche, trasporto a livello di membrane, trasmissione degli impulsi nervosi, contrazione muscolare, respirazione, circolazione, digestione, secrezione ormonale, attività biosintetiche, ecc);
- l’utilizzazione stessa dei nutrienti (termogenesi indotta dagli alimenti);
- la termo-regolazione;
- l’attività fisica.
Vi è pertanto la necessità di mantenere un continuo afflusso di nutrienti, in particolare di glucosio, ai tessuti. È fondamentale, quindi, che la concentrazione di glucosio nel sangue venga mantenuta entro un ristretto intervallo, in particolare per le esigenze del sistema nervoso, e ciò a fronte da un lato delle richieste continue e variabili da parte dei tessuti, e dall’altro della necessaria discontinuità dell’alimentazione.
Il controllo del metabolismo energetico è affidato soprattutto agli ormoni insulina, glucagone, adrenalina e cortisolo.
Subito dopo un pasto, giungono in circolo glucosio, acidi grassi e aminoacidi. L’aumento della glicemia stimola la secrezione di insulina da parte delle cellule β-pancreatiche delle insule di Langerhans. Il trasportatore di glucosio GLUT2 fa entrare il glucosio nella cellula β, dove viene convertito in glucosio 6-fosfato dall’esochinasi IV (gluco-chinasi) ed entra nel processo glicolitico. Ciò fa aumentare la concentrazione intra-cellulare di ATP, con conseguente chiusura dei canali per il K+ ATP-dipendenti della membrana plasmatica, ottameri formati da 4 subunità Kir 6.2 e 4 subunità SUR1 (recettori per le sulfoniluree). Il ridotto efflusso di K+ determina depolarizzazione della membrana, con conseguente apertura dei canali del Ca2+ e aumento del Ca2+ intra-cellulare che induce rilascio per esocitosi dell’insulina, rilascio stimolato anche dal parasimpatico, mentre il simpatico ha funzioni inibitorie.
L’insulina secreta in circolo si lega ai suoi recettori, inducendo la traslocazione sulla membrana plasmatica del trasportatore del glucosio GLUT4. Viene quindi aumentata l’assunzione di glucosio a livello:
- del tessuto muscolare, nel quale l’ormone stimola sia la glicolisi che la sintesi di glicogeno, mentre riduce la glicogenolisi;
- del tessuto adiposo, dove promuove la glicolisi e la sintesi di trigliceridi (attivazione della lipoprotein-lipasi).
Nel fegato l’insulina determina:
- aumento dell’espressione della gluco-chinasi, con conseguente aumento dell’assunzione di glucosio;
- attivazione della glicogeno-sintetasi e inibizione della glicogeno-fosforilasi;
- ossidazione del glucosio a piruvato (attivazione fosfofrutto-chinasi), che viene ossidato ad acetil-CoA (attivazione piruvato-deidrogenasi), che non viene ulteriormente ossidato nel ciclo di Krebs ma utilizzato per la sintesi degli acidi grassi (attivazione acetil-CoA carbossilasi) che, in forma di trigliceridi (VLDL), vengono esportati nel tessuto adiposo e nei muscoli.
Gli aminoacidi vengono utilizzati per la sintesi proteica nel fegato e negli altri tessuti, mentre gli aminoacidi in eccesso vengono convertiti in piruvato e in acetil-CoA, usato per la sintesi lipidica, che richiede anche NADPH derivante dal ciclo del pentosio-fosfato.
L’effetto dell’insulina è, pertanto, quello di trasformare i nutrienti e, in particolare il glucosio in eccesso, in forme di deposito dell’energia: glicogeno nel fegato e nel muscolo e trigliceridi nel tessuto adiposo.
Mentre la secrezione insulinica aumenta in risposta all’aumento della glicemia dovuto al pasto, la secrezione di glucagone ne risulta inibita.
A distanza di qualche ora dal pasto, i livelli di glucosio nel sangue tendono a ridursi per la continua utilizzazione da parte del cervello e degli altri tessuti. Si riduce, pertanto, la velocità della reazione esochinasica nelle cellule β-pancreatiche e, di conseguenza, il rilascio di insulina, mentre viene secreto glucagone dalle cellule α-pancreatiche. Il glucagone tende a ripristinare normali livelli glicemici, determinando aumento del rilascio di glucosio da parte del fegato, in conseguenza dell’attivazione della glicogeno-fosforilasi che demolisce il glicogeno, l’inattivazione della glicogeno-sintetasi, l’inibizione della glicolisi epatica e la stimolazione della gluconeogenesi che utilizza il piruvato, il lattato, il glicerolo derivante dai trigliceridi e gli aminoacidi glucogenici derivanti dal catabolismo proteico. A livello del tessuto adiposo il glucagone stimola la lipasi ormono-sensibile, che libera dai trigliceridi di deposito glicerolo (utilizzato dalla gluconeogenesi) e acidi grassi, che vengono ossidati dal fegato e da altri tessuti, con risparmio di glucosio, che può restare disponibile per il cervello. Tutti i processi controllati dal glucagone sono mediati dalla fosforilazione di proteine dipendente dal cAMP, mediatore intra-cellulare prodotto in seguito all’interazione del glucagone con il suo recettore.
Quando il digiuno si protrae e l’apporto di glucosio al cervello inizia a ridursi, i corpi chetonici prodotti dal fegato divengono una fonte di energia fondamentale per vari tessuti, compreso il cervello stesso (chetogenesi). Nel digiuno prolungato, pertanto, il fegato, per fornire glucosio al cervello, degrada alcune proteine e gli aminoacidi non essenziali che ne derivano vengono transaminati o deaminati: i gruppi aminici vengono trasformati in urea ed eliminati a livello renale, mentre gli scheletri carboniosi degli aminoacidi glucogenici vengono convertiti in piruvato o in intermedi del ciclo di Krebs e utilizzati, insieme al glicerolo derivante dalla degradazione dei trigliceridi, per produrre glucosio (gluconeogenesi). Gli acidi grassi vengono ossidati ad acetil-CoA, ma quando la concentrazione di ossalacetato si riduce in conseguenza dell’utilizzo di intermedi del ciclo di Krebs per la gluconeogenesi, l’acetil-CoA non può entrare nel ciclo di Krebs e si accumula, dando luogo alla formazione di acetoacetil-CoA e corpi chetonici nel fegato. Se il digiuno perdura a lungo, oltre alla deplezione delle riserve energetiche del tessuto adiposo (trigliceridi), si possono avere danni funzionali anche gravi a carico del cuore e del fegato, per la degradazione di proteine essenziali.
Anche l’adrenalina attiva la glicogeno-fosforilasi e inibisce la glicogeno-sintetasi, stimolando la liberazione di glucosio. Promuove anche l’utilizzazione anaerobica del glicogeno nel muscolo, fornendo energia di pronta utilizzazione (formazione glicolitica di ATP), con liberazione di lattato. Determina anche la mobilizzazione di acidi grassi liberi dal tessuto adiposo (attivazione della triacilglicerolo-lipasi). Stimola, inoltre, la secrezione di glucagone e inibisce la secrezione insulinica, favorendo la mobilizzazione di combustibili metabolici e inibendone il deposito. Ciò è importante, ad esempio, durante l’attività fisica, o quando l’animale viene a trovarsi nella situazione di stress che richiede una reazione immediata di attacco o fuga. In tali situazioni, la midollare del surrene libera adrenalina, mentre il sistema simpatico libera noradrenalina. Tali ormoni determinano dilatazione delle vie respiratorie per favorire l’arrivo di O2 ai tessuti e aumentano la pressione sanguigna, la frequenza cardiaca e l’afflusso di sangue ai muscoli, proprio per favorire la disponibilità dei substrati energetici liberati e la loro utilizzazione da parte dei tessuti, in particolare da parte del muscolo. Le azioni dell’adrenalina sono mediate dal cAMP, come per il glucagone. Anche in caso di ipoglicemia si ha liberazione di adrenalina e degli altri ormoni della contro-regolazione (glucagone, cortisolo e GH), con il compito di tentare di riportare la glicemia nel range di normalità. L’adrenalina è alla base della sintomatologia delle crisi ipoglicemiche (ansia, palpitazioni, tachicardia, tremore, pallore,…).
In varie situazioni di stress (digiuno, ipoglicemia, paura, ansia, dolore, emorragia, infezioni) aumenta la secrezione di cortisolo da parte della corticale del surrene. Il cortisolo stimola la scissione dei trigliceridi nel tessuto adiposo, determinando liberazione di acidi grassi, disponibili per l’ossidazione, e glicerolo, trasformato in glucosio dalla gluconeogenesi epatica. Il cortisolo stimola anche la degradazione di proteine muscolari, i cui aminoacidi glucogenici vanno anch’essi ad alimentare la gluconeogenesi epatica. Questa è attivata anche direttamente dal cortisolo che stimola la sintesi dell’enzima chiave, la fosfoenol-piruvato carbossi-chinasi. Il glucosio prodotto può essere utilizzato sia per far aumentare la glicemia, sia per la sintesi di glicogeno, necessario a supportare l’organismo nelle situazioni di stress prolungato.
Bibliografia
- Nelson DL, Cox MM. I principi di biochimica di Lehninger. 5° edizione. Zanichelli Editore.
Fisiologia delle incretine
Vincenzo Triggiani
Endocrinologia e Malattie Metaboliche, Dipartimento Interdisciplinare di Medicina, Università degli Studi di Bari “Aldo Moro”
L’osservazione che una quantità determinata di glucosio introdotta per via orale determinava una risposta insulinemica superiore rispetto a quella indotta dalla stessa quantità di glucosio somministrata per via endovenosa ha portato all’introduzione del concetto di asse entero-insulare e alla scoperta delle incretine, ormoni prodotti dall’intestino, che potenziano la secrezione insulinica in maniera glucosio-dipendente. Le più note sono il Glucagon-Like Peptide-1 (GLP-1) e il Glucose-dependent Insulinotropic Polipeptide (GIP).
Il GLP-1 è sintetizzato e secreto dalle cellule entero-endocrine L del tenue e del colon, per un processo post-traslazionale della prohormone convertase 1/3 (PC1/3) sulla molecola del pro-glucagone. GLP-1 è prodotto anche nel SNC (soprattutto nel tronco cerebrale), dove media effetti metabolici, cardiovascolari e di neuroprotezione.
Vi è una secrezione basale costante di GLP-1, che aumenta rapidamente dopo l’ingestione di nutrienti (carboidrati, grassi e proteine). La secrezione è in parte dipendente dalla produzione intestinale di chilomicroni e i livelli di GLP-1 sono più elevati nella linfa, nella quale passano i chilomicroni, che nel sangue portale. In tal modo parte del GLP-1 secreto non passa attraverso il filtro epatico.
Il GLP-1 stimola la secrezione di insulina e regola le concentrazioni di glucosio sia con azioni sul pancreas che extra-pancreatiche.
Il ruolo fisiologico del GLP-1 endogeno è stato studiato utilizzando modelli animali come i topi privi del recettore per il GLP-1 (GLP-1R-/-), nonché antagonisti del GLP-1R. Le azioni biologiche del GLP-1 possono così essere riassunte:
- riduzione della glicemia a digiuno;
- aumento della secrezione insulinica glucosio-dipendente;
- riduzione della secrezione di glucagone, con conseguente riduzione della gluconeogenesi epatica;
- riduzione dell’apoptosi delle ß-cellule pancreatiche;
- aumento della proliferazione e delle dimensioni delle ß-cellule pancreatiche;
- riduzione dell’introito di cibo;
- riduzione della spesa energetica;
- riduzione del peso corporeo;
- aumento del sensing portale del glucosio;
- aumento del metabolismo lipidico post-prandiale;
- aumento del controllo del flusso ematico da parte del SNC;
- miglioramento della funzione cardiovascolare;
- effetto neuroprotettivo.
Il GLP-1, pertanto, stimola in maniera glucosio-dipendente sintesi e secrezione di insulina, ripristina la sensibilità al glucosio nelle ß-cellule glucosio-resistenti, stimola proliferazione e neogenesi delle ß-cellule beta e ne inibisce l’apoptosi. Il GLP-1 inibisce la secrezione di glucagone da parte delle alfa-cellule sempre in modalità glucosio-dipendente. In presenza di concentrazioni di glucosio al di sotto di quelle fisiologiche, l’azione di stimolo sulla secrezione insulinica cessa.
Il GLP-1 agisce sulle ß-cellule insulari legandosi al proprio recettore, il GLP-1R, della famiglia dei recettori accoppiati a proteine G, attivando in tal modo, attraverso cAMP e protein-kinasi A, la via di segnale TCF7L2/Wnt- ß-catenina. La ß-arrestina 1 è richiesta per l’effetto sull’aumento della secrezione insulinica e per l’effetto citoprotettivo sulle ß-cellule, mentre l’effetto sulla sopravvivenza cellulare è legato all’aumento GLP-1 indotto dal rilascio di IGF-2, che agisce con meccanismo autocrino sulla stessa ß-cellula. Le azioni sulle alfa-cellule sono probabilmente indirette e mediate dalla somatostatina, la cui produzione da parte delle cellule delta del pancreas è aumentata per azione del GLP-1.
I nutrienti assorbiti a livello intestinale attivano un GLP-1R funzionale a livello della vena porta, innescando un circuito riflesso vagale, che controlla l’utilizzazione del glucosio da parte dell’organismo, indipendentemente dall’aumento della secrezione insulinica. Il glucosio assorbito a livello intestinale attiva, pertanto, network GLP-1R-sensibili nel SNC che promuovono l’utilizzazione di glucosio. Inoltre, il segnale del GLP-1R a livello cerebrale controlla il flusso ematico periferico e la sensibilità insulinica, prevalentemente in condizioni iperinsulinemiche-iperglicemiche.
Sui GLP-1R cerebrali agisce non solo il GLP-1 prodotto a livello cerebrale, ma anche il GLP-1 prodotto dalle cellule L-intestinali. Esso, infatti, supera la barriera emato-encefalica. Come detto in precedenza, inoltre, vi sono interazioni con il cervello mediate dai neuroni sensitivi afferenti vagali.
L’infusione ev di GLP-1 prima di un pasto ha determinato in volontari sani sensazione di sazietà e riduzione di circa il 12% delle calorie ingerite; inoltre, sia in soggetti sani che in diabetici tipo 2, determina un rallentamento dello svuotamento gastrico e un conseguente ritardo nell’assorbimento dei carboidrati.
La somministrazione di GLP-1 in infusione ev continua in diabetici tipo 2 (i quali hanno un “effetto incretinico” ridotto rispetto ai non diabetici) ha dimostrato di aumentare la secrezione insulinica e normalizzare sia la glicemia a digiuno che quella post-prandiale in soggetti in fallimento secondario con sulfaniluree. Il GLP-1 è in grado, inoltre, di ripristinare la prima fase della secrezione insulinica. Somministrato sc mediante micro-infusore per 6 settimane, ha determinato un netto miglioramento del profilo glicemico e una riduzione dell’emoglobina glicata, oltre a indurre riduzione dell’appetito e calo ponderale.
La somministrazione continua ev o sc di GLP-1 non è tuttavia proponibile, anche in relazione ai costi. La somministrazione sc di singole dosi in diabetici tipo 2 si è d’altro canto rivelata assolutamente deludente, in relazione all’emivita plasmatica molto breve di tale ormone, dell’ordine di pochi minuti, per l’intervento della dipeptidil-peptidasi-IV (DPP-IV), un enzima di membrana (ne esiste anche una forma circolante) presente in molti tessuti (rene, intestino, endotelio dei capillari, …), che scinde il dipeptide amino-terminale dei peptidi che presentano come penultimo aminoacido un residuo di prolina o di alanina. Sono state perseguite, pertanto, due diverse strategie per agire sul sistema incretinico nel soggetto con diabete tipo 2: 1) lo sviluppo di analoghi del GLP-1, agonisti del GLP-1R, a più lunga emivita, perché resistenti alla degradazione da parte della DPP-IV; 2) lo sviluppo di farmaci inibitori della DPP-IV, in grado cioè di prolungare l’emivita del GLP-1 endogeno.
I GLP-1 agonisti hanno molteplici effetti farmacologici:
- aumentano biosintesi e secrezione di insulina;
- promuovono la proliferazione e inibiscono l’apoptosi delle ß-cellule pancreatiche, anche se gli studi clinici non confermano che il trattamento con GLP-1 agonisti possa effettivamente aumentare la massa ß-cellulare funzionante nel diabetico;
- aumentano la secrezione di somatostatina e riducono quella di glucagone a livello pancreatico;
- riducono l’apporto di cibo;
- hanno effetti neuro-protettivi e promuoventi la neurogenesi a livello cerebrale, da cui derivano possibili implicazioni per il trattamento delle malattie neuro-degenerative;
- aumentano l’escrezione di sodio a livello renale, promuovendo diuresi e natriuresi;
- riducono l’infiammazione, soprattutto a livello cerebrale, pancreatico e cardiaco;
- aumentano l’utilizzazione di glucosio e riducono il metabolismo degli acidi grassi a livello cardiaco, migliorando la funzione cardiaca e determinando effetti cardio- e vaso-protettivi.
I GLP-1 agonisti, inoltre, hanno effetti indiretti, mediati dal cervello, su vari organi e apparati:
- riducono l’output di glucosio e lipidi da parte del fegato e riducono la steatosi epatica;
- riducono la lipogenesi nel tessuto adiposo bianco e, nell’animale, aumentano la termogenesi nel tessuto adiposo bruno;
- aumentano l’insulino-resistenza e riducono l’utilizzazione di glucosio da parte del muscolo;
- riducono lo svuotamento gastrico, aumentando il senso di sazietà;
- riducono la motilità intestinale.
In alcuni studi hanno dimostrato di poter ridurre il fabbisogno insulinico e di migliorare il controllo glico-metabolico nei diabetici tipo 1, sia C-peptide positivi che C-peptide negativi, anche se sono necessari ulteriori studi per determinare se la terapia con GLP-1 agonisti possa migliorare o conservare la funzione ß-cellulare nei diabetici di tipo 1 di nuova diagnosi.
Il GIP è un peptide di 42 aminoacidi, sintetizzato e secreto dalle cellule entero-endocrine K localizzate nel duodeno e nel digiuno prossimale, oltre che da aree del SNC (soprattutto ippocampo, bulbo olfattorio, ipotalamo, talamo e cervelletto).
In condizioni di digiuno, vi è una secrezione basale minima. I livelli plasmatici aumentano nell’arco di pochi minuti dopo l’ingestione di nutrienti.
Il recettore per il GIP (GIPR) appartiene alla famiglia di recettori accoppiati a proteine G. La sua attivazione determina produzione di cAMP e secrezione di insulina da parte delle ß-cellule.
Anche il GIP è degradato dall’enzima DPP-IV, che rappresenta, pertanto, il principale regolatore della degradazione delle incretine.
Il GIP:
- stimola la sintesi e la secrezione insulinica con modalità glucosio-dipendenti;
- aumenta la secrezione di glucagone;
- esercita numerose azioni extra-pancreatiche a livello di tratto gastro-intestinale, tessuto adiposo, cuore, ipofisi, cervello, osso e corticale del surrene;
- promuove la captazione di lipidi, la lipogenesi e l’espansione della massa adipocitaria, con aumento di secrezione di adipochine;
- aumenta il metabolismo dei trigliceridi a livello cardiaco;
- aumenta la formazione e riduce il riassorbimento a livello osseo;
- ha effetti neuro-protettivi e favorenti la neurogenesi, oltre ad agire sull’endotelio;
- svolge azioni anti-apoptotiche su ß-cellule insulari in vitro.
Il trattamento con GIP esogeno non è in grado di stimolare la secrezione insulinica nella maggior parte dei soggetti iperglicemici con diabete tipo 2, mentre 4 settimane di trattamento insulinico non solo migliorano il controllo glico-metabolico, ma ripristinano la risposta insulinotropa al GIP esogeno in tali soggetti.
Bibliografia essenziale
- Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab 2013, 17: 819-37.
- Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev 2012, 33: 187-215.
Diagnostica biochimica per il diabete
Glicemia e glicosuria
Emoglobina glicata
Insulina, proinsulina e peptide C
Lipidi
Chetoni
Albuminuria
Test dinamici
Glicemia e glicosuria nella diagnostica del diabete
Achiropita Pucci
Endocrinologia territoriale, ASP Cosenza
Il glucosio nel sangue può essere misurato sia su plasma (prelievo venoso) sia su sangue intero (prelievo capillare).
Il prelievo plasmatico della glicemia è d'importanza fondamentale per la diagnosi e il monitoraggio del diabete mellito di I e II tipo.
Il rilievo della glicemia su sangue capillare, da polpastrello o da altri, meno comuni, siti alternativi (braccio, palmo della mano, lobo dell’orecchio), effettuato con glucometri a determinazione quantitativa elettrochimica, è un utile strumento di auto-monitoraggio del diabete ma non può essere usato a fini diagnostici.
DIAGNOSI
Il livello della glicemia plasmatica dopo 8 ore di digiuno deve essere < 100 mg/dL. Valori superiori (iperglicemia a digiuno o impaired fasting glucose o IFG), soprattutto in soggetti con fattori di rischio per malattia diabetica (familiarità, sovrappeso o obesità, età > 45 anni, segni di insulino-resistenza), rappresentano sempre un campanello d'allarme e inducono la necessità di eseguire una curva da carico con 75 g di glucosio a 120 minuti. In base al risultato di questo test dopo 2 ore, si possono distinguere tre categorie:
- glicemia < 140 mg/dL: normale;
- glicemia tra 140 e 199 mg/dL: ridotta tolleranza glucidica o impaired glucose tolerance o IGT;
- glicemia ≥ 200 mg/dL: diabete mellito.
Il riscontro di valori di glicemia plasmatica a digiuno ≥ 126 mg/dL, confermato da una seconda determinazione della glicemia a digiuno, ci permette di porre diagnosi di diabete mellito.
Anche il rilievo casuale di una glicemia > 200 mg/dL, indipendentemente dall’assunzione di cibo e in presenza di sintomi di malattia diabetica (poliuria, polidipsia, polifagia, dimagramento), consente la diagnosi di diabete.
Raccomandazioni
- La diagnosi di diabete richiede la misurazione del glucosio nel plasma. Livello e qualità di evidenza: A, alta
- Il sangue per l'analisi del glucosio plasmatico deve essere prelevato dopo almeno 8 ore di digiuno. Se il plasma non può essere separato dalle cellule entro 60 minuti, il sangue deve essere raccolto in una provetta contenente un inibitore della glicolisi, come il fluoruro di sodio. Livello e qualità di evidenza: B, moderata.
MONITORAGGIO
I prelievi di glicemia capillare sono indispensabili per il monitoraggio del diabete, soprattutto se insulino-trattato. I dati pre- e post-prandiali di glicemia rilevati attraverso i glucometri permettono di verificare il raggiungimento dei target glicemici individuali e prefissati e, quindi di monitorare l'andamento giornaliero della glicemia in relazione a pasti, attività fisica, patologie acute, terapia farmacologica.
I valori della glicemia capillare risultano inferiori a quelli della glicemia plasmatica e il dato di accuratezza e precisione varia dal 4 al 20% (coefficiente di variabilità), a secondo del tipo di glucometro utilizzato; gli strumenti di ultima generazione, plasma-calibrati, rilevano, tuttavia, valori che si discostano poco dai dati di laboratorio.
I dati dell'autocontrollo glicemico, correlati ai valori di HbA1c, rappresentano un indispensabile strumento per apportare modifiche terapeutiche, utili al fine del raggiungimento del compenso glicemico ottimale. Gli obiettivi glicemici da perseguire sono:
- glicemia a digiuno e pre-prandiale compresa tra 70 e 130 mg/dL;
- glicemia post-prandiale < 180 mg/dL.
| Tabella 1 Alterazioni delle concentrazioni di glucosio da cause fisiopatologiche |
|
| Diminuite da | Aumentato consumo (digiuno, attività fisica, eccesso di insulina) Ridotta produzione endogena (insufficienza epatica, galattosemia) Glicosuria renale |
| Aumentate da | Obesità Stress (infarto del miocardio, danno cerebrale, convulsioni, trauma, anestesia generale) Sindrome di Cushing Feocromocitoma Acromegalia Epatopatia cronica Pancreatite Ipertiroidismo |
| Tabella 2 Modificazioni farmaco-indotte delle concentrazioni di glucosio |
|
| Diminuite da | Salicilati Chinina Anti-tubercolari Ipoglicemizzanti Insulina |
| Aumentate da | Tiazidi e altri diuretici Cortisonici Glucagone |
Il razionale di questa determinazione è che la concentrazione di glucosio nelle urine riflette la concentrazione media di glucosio nel sangue durante il periodo della raccolta che eccede la capacità di riassorbimento del tubulo renale (mediamente 180 mg/dL), nel caso che la raccolta si riferisca a un determinato periodo di tempo (di solito da 4 a 24 ore), mentre il campione estemporaneo di urine riflette la concentrazione di glucosio al momento della raccolta.
Oggi si consiglia l'automonitoraggio del glucosio nelle urine solo per quei pazienti che non sono in grado o non vogliono eseguire l'automonitoraggio nel sangue (numero non irrilevante). I pazienti e, naturalmente, i medici che valutano i risultati devono avere ben chiaro che le determinazioni su urine non forniscono nessuna informazione circa concentrazioni di glucosio circolante inferiori alla soglia renale.
I motivi per cui l'autodeterminazione nelle urine è meno consigliabile rispetto a quella nel sangue sono i seguenti:
- il valore di soglia renale per il glucosio presenta ampie oscillazioni e spesso si discosta dalla concentrazione classica di 10 mmol/L (180 mg/dL). Questo è rilevante in molti casi, per esempio negli adulti con diabete mellito da lungo periodo, in cui la soglia può aumentare anche in modo notevole (portando quindi a una sottostima della concentrazione di glucosio nel sangue), nei bambini e, in particolare, nelle donne in gravidanza che possono avere una soglia renale molto bassa o variabile;
- l'assunzione di liquidi e la concentrazione delle urine può influenzare la determinazione di glucosio nelle urine;
- la concentrazione di glucosio nelle urine riflette la concentrazione media di glucosio nel sangue a partire dall'ultima minzione e non al momento della raccolta;
- non rilevare glucosio nelle urine non discrimina tra ipoglicemia, euglicemia e iperglicemia lieve o moderata; questa analisi non è pertanto utile per prevenire ipo- ed iperglicemia;
- la determinazione del glucosio che si basa sul confronto visivo con una carta di colori è meno accurata della misurazione nel sangue capillare, che di solito si avvale della lettura reflettometrica digitale;
- la determinazione del glucosio nelle urine soffre di interferenze da alcuni farmaci (il glucosio è aumentato da carbamazepina, corticosteroidi, d-tiroxina, diuretici - ad esempio acetazolamide, acido etacrinico, furosemide, tiazidici - EDTA, litio carbonato, acido nicotinico).
BIBLIOGRAFIA
- American Diabetes Association. Standards of medical care in diabetes-2006. Diabetes Care 2006, 29 (suppl 1): S4-S42.
- AMD-SID. Standard Italiani per la cura del Diabete mellito. 2014.
Emoglobina glicata
Achiropita Pucci
Endocrinologia territoriale, ASP Cosenza
L'emoglobina glicata (HbA1c) è il prodotto finale della glicazione dei residui aminici della proteina contenuta nei globuli rossi ed è in rapporto proporzionale alla concentrazione media di glucosio presente nel sangue. Considerata la vita media di 90 giorni dei globuli rossi, la percentuale di glicazione dell'emoglobina rappresenta un indice medio della quantità di glucosio circolante, relativo ai 2-3 mesi antecedenti al prelievo: non è, pertanto, in grado di rivelare grandi variazioni di concentrazioni di glucosio se non dopo molte settimane. L'HbA1c è una misura "pesata" della concentrazione media di glucosio. Il risultato finale è influenzato più dal passato recente che da quello remoto: la concentrazione media del mese precedente contribuisce per circa il 50% al risultato finale, mentre quella nel periodo compreso tra il 90° e il 120° giorno precedente il prelievo "pesa" per circa il 10%.
Naturalmente qualunque condizione alteri il turn-over dei globuli rossi (anemia, emorragia, ecc) modifica il risultato finale dell'emoglobina glicata e non la correla più con precisione alla quantità di glucosio presente nel sangue (tab. 1)
| Tabella 1 Scala di correlazione fra HbA1c e glicemia media |
||
| HbA1c (%) | Glicemia media (mmol/L) | Glicemia media (mg/dL) |
| 5 | 4.5 | 90 |
| 6 | 6.7 | 120 |
| 7 | 8.3 | 150 |
| 8 | 10.0 | 180 |
| 9 | 11.6 | 210 |
| 10 | 13.3 | 240 |
| 11 | 15.0 | 270 |
| 12 | 16.7 | 300 |
L'HbA1c è diventata, nella pratica clinica, il principale indice di valutazione del compenso metabolico glicidico e se ne consiglia il dosaggio ogni 3-6 mesi a secondo della tipologia del paziente e dell’andamento della malattia diabetica. Viene, invece, meno usata a scopo diagnostico.
Diversi studi scientifici hanno dimostrato e confermato che l'aumento di HbA1c è direttamente proporzionale all'incremento del rischio cardiovascolare e hanno portato alla definizione del target da perseguire per ridurre l'incidenza di malattie cardiovascolari.
Attualmente è in uso la misurazione dell'HbA1c in mmol/mol anziché in %, secondo una metodica standardizzata (DDCT) allineata con il sistema internazionale di unità di misura (tab. 2).
| Tabella 2 Corrispondenza valori HbA1c nelle 2 unità di misura |
|
| DCCT (%) | IFCC (mmol/mol) |
| 4.0 | 20 |
| 5.0 | 31 |
| 6.0 | 42 |
| 6.5 | 48 |
| 7.0 | 53 |
| 7.5 | 59 |
| 8.0 | 64 |
| 9.0 | 75 |
| 10.0 | 86 |
| 11.0 | 97 |
| 12.0 | 108 |
Nei soggetti non diabetici l'HbA1c è sempre < 6.5% (DCCT) o < 42 mmol/mol (IFCC).
In soggetti diabetici, valori di HbA1c < 6.5-7% (DCCT) o < 48-53 mmol/mol (IFCC) sono indici di buon compenso metabolico e rappresentano il target perseguibile in pazienti non fragili, per età e patologie correlate. Il valore di HbA1c deve, comunque, sempre essere correlato ai dati dell'autocontrollo glicemico effettuato dal paziente su sangue capillare: ciò permette di valutare le variabilità glicemiche e le ipoglicemie, di grande peso nel buon controllo nella malattia diabetica.
| Tabella 3 Alterazioni delle concentrazioni di HbA1c da cause fisiopatologiche |
|
| Diminuite da | Gravidanza Insufficienza renale - uremia (alcuni metodi) Ogni condizione che aumenta il turn-over degli eritrociti ed arricchisce il pool con cellule più giovani:
|
| Aumentate da | Leggermente con l’età (0.03% per anno) Ogni condizione che aumenta la sopravvivenza degli eritrociti:
Emoglobina F (HPLC, elettroforesi) |
Esistono poi differenze etniche (≤ 0.4%, dovute a fattori quali la cinetica di ingresso del glucosio negli eritrociti e variazioni della vita media eritrocitaria) e stagionali (fino a circa il 7%, di tipo ciclico, con periodo semestrale).
| Tabella 4 Modificazioni farmaco-indotte delle concentrazioni di HbA1c |
|
| Diminuite da | Assunzione di vitamina C o E (almeno 1 g/die) |
| Aumentate da | Acido acetil-salicilico (HPLC, elettroforesi) Alcool (HPLC, elettroforesi) Dipendenza da oppiacei |
Bibliografia
- AMD-SID. Standard Italiani per la cura del diabete mellito. 2014.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352: 837-53.
- The ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008, 358: 2560-72.
- Duckworth W, Abraira C, Moritz T, et al; the VADT Investigators. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med 2009, 360: 129-39.
- DECODE Study Group, European Diabetes Epidemiology Group. Is the current definition for diabetes relevant to mortality risk from all causes and cardiovascular and noncardiovascular diseases? Diabetes Care 2003, 26: 688-96.
Insulina, proinsulina e peptide C nella diagnostica del diabete
Isabella Romano
Endocrinologia e Malattie del Ricambio, Cittadella della Salute, ASL Lecce
L’insulina è un ormone essenziale per mantenere l’omeostasi metabolica dell’organismo. Per produrre l’insulina biologicamente attiva, la ß-cellula pancreatica inizia la sintesi partendo da un precursore, la pre-proinsulina. Questa è una proteina dal peso molecolare di 11.500 Dalton, sintetizzata nel reticolo endoplasmatico rugoso della ß-cellula, che, per delezione enzimatica di circa 20 aminoacidi dal terminale aminico, si trasforma in pro-insulina, proteina a catena semplice composta da 86 aminoacidi. La pro-insulina, attraverso i canali del reticolo endoplasmatico, viene trasportata all’apparato di Golgi in granuli secretori immaturi, nei quali avviene una seconda scissione enzimatica, con perdita di quattro aminoacidi e formazione di insulina biologicamente attiva e di un peptide di coniugazione costituito da 31 aminoacidi, detto Peptide C.
L’insulina è costituita da 51 aminoacidi, divisi in due catene polipeptidiche (subunità A e B) legate da due ponti disolfuro (un terzo ponte disolfuro è presente anche all’interno della catena A). La subunità A è costituita da 21 aminoacidi e quella B da 30 aminoacidi. Dopo il taglio proteolitico della pro-insulina, peptide C e insulina vengono immagazzinati, in quantità equimolare, nei granuli maturi pronti per la secrezione in circolo. Una minima parte di pro-insulina può essere secreta in circolo intatta, ma è biologicamente meno attiva dell’insulina. Insulina e peptide C invece vengono regolarmente secreti dalla ß-cellula pancreatica in quantità equimolari (una molecola di C-peptide e una di insulina).
Il peptide C, al contrario dell’insulina, non viene metabolizzato a livello epatico, ma renale e viene escreto con le urine. Pertanto, in virtù della maggiore emivita, la concentrazione plasmatica di peptide C è superiore a quella dell’insulina, anche se secreto dal pancreas in modo equimolare.
Da quanto esposto, si comprende come pro-insulina, insulina e peptide C possano rappresentare utili parametri di riferimento laboratoristici per la valutazione del paziente diabetico.
I metodi di misura per insulina, pro-insulina e peptide-C hanno due applicazioni principali: la valutazione dell’ipoglicemia e quella, essenzialmente di ricerca, relativa alla patogenesi e alla terapia del diabete.
In ambito clinico si devono valutare due aspetti:
- se l’ipoglicemia è associata a una soppressione appropriata della (pro)insulina;
- se l’inappropriata elevazione dell’insulinemia è endogena (vale a dire accompagnata da un aumento della concentrazione di peptide-C).
Da molti anni è noto che le cellule adenomatose dell’insula producono pro-insulina e una quantità variabile di insulina e che la concentrazione di pro-insulina circolante può variare nei diversi pazienti. Quasi il 90% dei pazienti con insulinoma presenta una quota di Pro-insulin Like Component (PLC) superiore al 25% del totale dell’insulina immuno-reattiva. È stato sostenuto, addirittura, che con l’uso di metodi specifici per la determinazione dell’insulina si potrebbe mancare la diagnosi di un insulinoma che produca soprattutto pro-insulina e che, analogamente, la somministrazione factitia di insulina animale potrebbe non essere rivelata. È da tenere presente anche il particolare rapporto che esiste tra insulina e glucosio nel neonato, a termine e prematuro, che presenta un’elevazione significativa di pro-insulina.
Pro-insulina
Il livello di pro-insulina plasmatica è un predittore di diabete mellito tipo 2, obesità e malattie cardiovascolari. Il rapporto tra pro-insulina e insulina (PI/I ratio) riflette la disfunzione della ß-cellula associata con l’inizio e la progressione del diabete mellito 2. Un elevato rapporto PI/I è attribuibile a un aumento della richiesta secretoria delle ß-cellule correlato con obesità e/o insulino resistenza. L’aumento della richiesta secretoria può determinare secrezione di granuli immaturi da parte delle ß-cellule, con aumento del contenuto di pro-insulina intatta.
Insulina
Il dosaggio dell’insulina endogena riveste un particolare interesse per valutare la persistenza della secrezione ß-cellulare. Nel diabete tipo 1 i valori insulinemici basali sono marcatamente ridotti, con incremento pressoché assente, dopo i pasti o dopo stimolo con glucosio. Nel diabete tipo 2 la risposta insulinemica è per lo più ritardata, anche se persistente. In presenza di una condizione di obesità, i livelli assoluti di insulina dopo uno stimolo glucidico possono presentare valori superiori a quelli riscontrabili nei soggetti normali nella fase tardiva della curva. Il dosaggio dell’insulinemia, unitamente a quello dei livelli ematici di peptide C, aiuta a valutare il contributo dell’insulina endogena, quindi prodotta dall’organismo e di quella somministrata dall’esterno (esogena).
| Tabella 1 Cause che possono modificare i livelli di insulinemia |
|
| Aumentati | Diabete mellito tipo 2 all’esordio Insulino-resistenza Alterata tolleranza al glucosio Insulinoma Acromegalia Sindrome di Cushing Farmaci corticosteroidi Contraccettivi orali |
| Diminuiti | Diabete mellito tipo 1 Ipopituitarismo Malattie pancreatiche: pancreatite, tumori del pancreas |
Peptide C
Anche se la concentrazione plasmatica del peptide-C non è uguale in termini molari a quella dell’insulina (in virtù della maggiore emivita del peptide-C), riflette comunque la capacità di sintesi di insulina endogena. Pertanto, nei pazienti con diabete tipo 1 i livelli di peptide-C nel sangue e nelle urine sono molto bassi. In questi pazienti la misurazione del peptide-C plasmatico viene spesso effettuata a scopo clinico, per valutare quale sia la produzione residua di insulina endogena, dal momento che quella esogena è priva del peptide di coniugazione. Nei pazienti con diabete di tipo 2, in cui l’alterazione prevalente è la resistenza periferica all’insulina che spesso si accoppia ad alterata secrezione insulinica, i livelli di peptide-C sono normali o superiori alla norma. Anche in questi pazienti la secrezione di peptide-C segue quella dell’insulina e si accoppiano iperinsulinemia e concentrazioni di C-peptide normali o aumentate.
Il dosaggio ematico di peptide-C è utile nella diagnosi di insulinoma, tumore che colpisce le ß-cellule pancreatiche provocando iperinsulinemia associata a ipoglicemia ed elevati livelli di peptide C nel sangue.
La valutazione della funzione ß-cellulare mediante misurazione del C-peptide basale o dopo stimolo con glucagone è utile nel sospetto clinico di LADA. Il test è utile per l’inquadramento diagnostico e prognostico dei casi di incerta classificazione, ma non rappresenta l’unico criterio su cui basare la scelta terapeutica.
Da alcuni anni una serie di studi ha suggerito che il peptide-C sia un ormone indipendente dall’insulina, con una propria funzione biologica. L’ipotesi che viene avanzata attualmente è che il peptide-C, modulando vie intra-cellulari non ancora completamente identificate, abbia un’azione protettiva nei confronti delle complicanze del diabete, in particolare della nefropatia e della neuropatia e successivamente anche della microangiopatia diabetica, caratterizzate da un decorso clinico più moderato.
| Tabella 2 Cause che possono modificare i livelli di peptide-C |
|
| Aumentati | Diabete mellito tipo 2 Pre-diabete Obesità Sindrome dell’ovaio policistico Sindrome di Cushing Insulinoma Eccessivo utilizzo di ipoglicemizzanti orali |
| Diminuiti | Diabete mellito tipo 1 LADA Diabete mellito tipo 2 in fase avanzata Malattie epatiche Infezioni gravi Morbo di Addison Iniezione di una dose eccessiva di insulina |
Bibliografia
- Liu M, Wright J,Guo H, et al. Proinsulin entry and transit through the endoplasmic reticulum in pancreatic beta cells. Vitam Horm 2014, 95: 35-62.
- Pfutzner A, Pfutzner AH, Larbig M, Forst T. Role of intact proinsulin in diagnosis and treatment of type 2 diabetes mellitus. Diabetes Technol Ther 2004, 6: 405-12.
- SID-AMD. Standard italiani per la cura del diabete mellito. 2014.
- Martina V, Maestroni A, Maestroni S, Zerbini G. Role of C-peptide in the pathogenesis of diabetic nefropathy. G It Nefrol 2010, 3: 240-8.
- Walenciak L, Fendler W, Mlynarski W. Proinsulin C-peptide-the bioactive peptide with a huge promise. Pediatr Endocrinol Diabetes Metab 2007, 13: 95-8.
Lipidi nella diagnostica del diabete
Paola Romagni
Specialista in Endocrinologia e Malattie del Ricambio
UOC di Medicina Interna - ASL Teramo
La “dislipidemia diabetica” è tipicamente caratterizzata dall’aumento di trigliceridi, LDL, HDL più piccole e dense e lipoproteine ricche di trigliceridi in fase post-prandiale (tabella) (1-4). Per valutare il rischio cardiovascolare nel paziente diabetico, sono stati proposti anche altri indici, quali il colesterolo non HDL e, più recentemente, il rapporto apoB/apoA1 (rischio elevato: uomini > 0.9; donne > 0.8), anche se i costi elevati e i problemi di standardizzazione delle metodiche rendono ancora difficile l’utilizzo di tali parametri nella pratica clinica.
| Assetto lipidico caratteristico del soggetto diabetico che contribuisce all’ulteriore aumento del rischio cardiovascolare | |
| Trigliceridi e lipoproteine ricche di trigliceridi | ↑ |
| HDL-C | ↓ |
| Trigliceridi post-prandiali | ↑ |
| Apo-A1 | ↓ |
| HDL piccole, preß1-HDL, a3-HDL | ↓ |
| Apo-B | ↑ |
| LDL | ↑ |
| LDL piccole e dense | ↑ |
| Apo-C-III | ↑ |
| Non-HDL-C | ↑ |
| Lipidi ossidati e glicosilati | ↑ |
Il colesterolo LDL rimane, quindi, il principale fattore di rischio cardiovascolare anche nel paziente diabetico, nonostante la nota 13 non ne preveda la rimborsabilità. Un’opzione per la valutazione indiretta di LDL colesterolo è l’applicazione della formula di Friedewald:
LDL = colesterolo totale - (HDL + trigliceridi/5)
Il colesterolo LDL calcolato con la formula di Friedewald sembrerebbe generalmente sottostimare il valore di LDL misurato direttamente. Tale dato è stato confermato in un recente studio che ha dimostrato come LDL colesterolo misurato direttamente sia generalmente maggiore di 5 mg/dL o del 5% rispetto al colesterolo LDL calcolato (5). Tale differenza risulterebbe maggiore nei pazienti con DM o in terapia con statina. Anche se alcuni studi hanno dimostrato una limitazione di tale metodo in pazienti con ipertrigliceridemia (> 400 mg/dL) e malattie epato-renali, la formula di Friedewald rappresenterebbe un buon metodo nei pazienti con sindrome metabolica (5-6).
Il controllo del profilo lipidico completo (colesterolo totale, HDL, trigliceridi) andrebbe effettuato almeno annualmente e ad intervalli di tempo più ravvicinati in caso di mancato raggiungimento dell’obiettivo terapeutico.
Il colesterolo LDL deve essere considerato l’obiettivo primario della terapia e l’obiettivo terapeutico da raggiungere è rappresentato da valori < 200 mg/dL. Possono essere valutati anche i livelli di apoB (target < 200 mg/dL), tenendo però presente che ci sono dei costi aggiuntivi e che le metodiche di dosaggio non sono ancora uniformemente standardizzate.
Bibliografia
- Canadian Diabetes Association clinical practice guidelines expert committee. Dyslipidemia. Can J Diabetes 2013, 37: S110-6.
- Fruchart JC, et al. The residual risk reduction iniziative: a call to action to reduce residual vascular risk in dyslipidemic patients. Diabetes Vasc Dis Res 2008, 5: 319-32.
- Parhofer KG, et al. Pathophysiology of diabetic dyslipidemia: implications for atherogenesis and treatment. Clin Lipidol 2011, 6: 401-11.
- AMD-SID. Standard italiani per la cura del diabete mellito. 2014.
- Choi SY, et al. Difference between calculated and direct-measured low-density lipoprotein cholesterol in subjects with diabetes mellitus or taking lipid-lowering medications. J Clin Lipidol 2012, 6: 114-20.
- Knopfholz J, et al. Validation of the Friedewald formula in patients with metabolic syndrome. Cholesterol 2014, 2014: 261878.
Chetoni
Paola Romagni
Specialista in Endocrinologia e Malattie del Ricambio
UOC di Medicina Interna - ASL Teramo
I corpi chetonici aceto-acetato (AcAc), acetone e ß-idrossi-butirrato (ßHBA) sono prodotti del catabolismo degli acidi grassi liberi. L’acetone, presente solo in piccole quantità, deriva dalla decarbossilazione spontanea dell’AcAc.
ßHBA e AcAc sono di solito presenti in quantità equimolari, ma il loro equilibrio è spostato verso la formazione di ßHBA in tutte le situazioni che alterano lo stato ossido-riduttivo dei mitocondri epatici, come ipossia, digiuno, disordini metabolici e cheto-acidosi alcolica.
L’acetone è usualmente presente in modeste quantità quale spontanea decarbossilazione dell’AcAc.
I metodi di determinazione dei corpi chetonici che non comprendono la determinazione del ßHBA possono, quindi, fornire informazioni fuorvianti, sottostimando la concentrazione dei corpi chetonici totali.
I chetoni sono normalmente presenti nelle urine, ma in concentrazioni inferiori alla sensibilità dei comuni metodi di dosaggio. La chetonuria è rilevabile negli individui normali durante il digiuno e fino al 30% dei campioni urinari del primo mattino della donna in gravidanza (con o senza diabete), o anche dopo episodi ipoglicemici.
Risultati falsamente positivi sono possibili con i test che utilizzano reagenti contenenti nitro-prussiato, in presenza di farmaci con gruppi sulfidrilici, quali l’ACE-inibitore captopril. Risultati falsamente negativi sono stati descritti con l’impiego di materiali inappropriatamente conservati, in presenza di urine fortemente acidificate (come dopo assunzione di elevate dosi di acido ascorbico) o in campioni ad elevata attività microbica capace di indurre consumo dei chetoni.
In assenza di glucosio, i corpi chetonici, il cui substrato è costituito prevalentemente dagli acidi grassi presenti nel tessuto adiposo e nel fegato, forniscono la maggior parte dell’energia necessaria all’organismo. Nel diabete non controllato, la bassa concentrazione di insulina aumenta la lipolisi e diminuisce la riesterificazione, aumentando la concentrazione di acidi grassi plasmatici. L’aumento degli ormoni contro-regolatori aumenta la lipolisi nel tessuto adiposo e la chetogenesi nel fegato. L’aumentata produzione epatica di chetoni e il diminuito metabolismo periferico portano ad accumulo di AcAc nel sangue: una piccola frazione viene spontaneamente decarbossilata ad acetone, ma la maggioranza è convertita a ßHBA, la cui concentrazione è maggiore su base molare.
La determinazione dei chetoni nelle urine (e nel sangue), facilmente misurabili sia in ambiente ospedaliero che domiciliare/ambulatoriale, è ampiamente utilizzata nella cura del paziente diabetico, sia per la diagnosi che nel monitoraggio della chetoacidosi diabetica. Il test per i chetoni urinari è importante anche per il monitoraggio del paziente diabetico, soprattutto nei soggetti con diabete di tipo 1, in corso di gravidanza con diabete pre-esistente e nel diabete gestazionale. Tutti i diabetici dovrebbero eseguire il test per la chetonuria durante qualunque tipo di malattia intercorrente, quando sono presenti intensa glicosuria o marcata e persistente iperglicemia (> 300 mg/dL), durante la gravidanza, o in presenza di sintomi compatibili con la diagnosi di chetoacidosi (nausea, vomito, dolore addominale).
Bibliografia
- Sacks DB, et al. Guidelines and recommendations for laboratory analysis in the diagnosis and management of diabetes mellitus. Clin Chem 2002, 48: 436-72.
- ADA. Standards of medical care in diabetes-2007. Diabetes Care 2007, 30: S4-41.
Albuminuria
Paola Romagni
Specialista in Endocrinologia e Malattie del Ricambio
UOC di Medicina Interna - ASL Teramo
La microalbuminuria è a tutt’oggi il più semplice e sensibile parametro per rilevare il rischio di nefropatia nel diabete mellito. Sia nel diabete mellito tipo 1 che tipo 2, ne sono stati riconosciuti e confermati il valore predittivo di nefropatia, uremia, morbilità e mortalità precoci, soprattutto per cause cardiovascolari.
Dal punto di vista fisiopatologico, nella nefropatia diabetica una maggiore quantità di albumina può attraversare la membrana basale glomerulare a causa della perdita precoce di proteoglicano anionico eparan-solfato. Le dimensioni dell’albumina sono sufficientemente piccole da consentirle di attraversare tale membrana, ma sufficientemente grandi da impedirle di essere riassorbita dal tubulo renale con una risultante proteinuria selettiva. Quando l’escrezione di albumina supera i 300 mg/die (macroalbuminuria), la filtrazione glomerulare aumenta di circa 1 mL/min per mese. Con il progredire della nefropatia, la proteinuria diventa non selettiva e le proteine urinarie acquistano la stessa composizione e distribuzione di quelle del siero.
L’escrezione urinaria dell’albumina può essere influenzata da fisiologiche variazioni circadiane, da diverse situazioni fisiopatologiche (tabella 1) e dall’assunzione intercorrente di farmaci (tabella 2).
| Tabella 1 Cause fisiopatologiche di albuminuria transitoria |
| Esercizio fisico intenso Infezioni delle vie urinarie Malattia febbrile Scompenso cardiaco congestizio Mestruazioni Iperglicemia transitoria Ipertensione grave |
| Tabella 2 Modificazioni iatrogene dell’albuminuria |
|
| Diminuita da | ACE-inibitori Sartani |
| Aumentata da | Amikacina Teofillina Sulfametossazolo+trimetoprim Venlafaxina Paromomicina Colimicina |
La microalbuminuria è definita come l’escrezione di piccole concentrazioni di albumina (30-299 mg/g o µg/mg creatinina o 20-199 µg/min o 30-299 mg/24h) (tabella 3), rilevata in almeno due su tre raccolte eseguite ad intervalli di 3-6 mesi, dopo che siano state escluse le condizioni che ne riducono l’affidabilità.
La raccolta urine/24 ore rimane il gold standard, sebbene sia una metodica abbastanza indaginosa e suscettibile di errori correlati alle modalità di raccolta in pazienti spesso anziani e scarsamente collaboranti (1-3). Un’alternativa è rappresentata dalla raccolta minutata, che però risente maggiormente della concentrazione delle urine. La raccolta spot, in cui si misura il rapporto albumina/creatinina urinaria (ACR), risulta sufficientemente predittiva dei valori di albuminuria/24h e rappresenta quindi un’alternativa valida e di più facile esecuzione (2-4). I cut-off per l’interpretazione dei valori di albuminuria sono schematizzati nella tabella 3.
| Tabella 3 Escrezione urinaria di albumina (4) |
|||
| Categoria | Raccolta spot (mg/g o µg/mg creatinina) |
Raccolta minutata (µg/min) |
Raccolta nelle 24 ore (mg/24 ore) |
| Albuminuria normale | < 10 (uomini) < 15 (donne) |
< 10 | < 10 |
| Albuminuria alta-normale | < 25 (uomini) < 35 (donne) |
10-19 | 10-29 |
| Microalbuminuria | 30-299 | 20-199 | 30-299 |
| Macroalbuminuria | ≥ 300 | ≥ 200 | ≥ 300 |
Lo screening per valutare l’escrezione renale di albumina ed escludere la presenza di albuminuria dovrebbe essere effettuato:
- nei diabetici di tipo 1 annualmente dopo i primi 5 anni dalla diagnosi;
- nei diabetici di tipo 2 e nelle donne diabetiche in gravidanza alla diagnosi e poi annualmente.
In tutti gli adulti diabetici, indipendentemente dal grado d’escrezione urinaria di albumina, dovrebbe essere misurata annualmente la creatininemia. Questa non dovrebbe essere usata da sola come misura della funzionalità renale, ma piuttosto essere utilizzata per stimare la velocità di filtrazione glomerulare, per eseguire una stadiazione della malattia.
Per la metodica di dosaggio: vedi.
Bibliografia
- Remuzzi G, et al. Nephropathy in patients with type 2 diabetes. N Engl J Med 2002, 346: 1145-51.
- Canadian Diabetes Association clinical practice guidelines expert committee. Chronic kidney disease in diabetes. Can J Diabetes 2013, 37: S129-3.
- Bakker AJ, et al. Detection of microalbuminuria. Receiver operating characteristic curve analysis favors albumin-to-creatinine ratio over albumin concentration. Diabetes Care 1999, 22: 307-13.
- AMD-SID. Standard italiani per la cura del diabete mellito. 2014.
Test dinamici per lo studio delle alterazioni del metabolismo glucidico
OGTT nella diagnostica delle alterazioni del metabolismo glucidico
Gabriele Zardini
UOC Medicina Generale ad indirizzo Endocrinologico, Azienda Ospedaliera Universitaria Integrata di Verona
| OGTT nella diagnostica delle alterazioni del metabolismo glucidico | |
| Indicazioni |
È uno dei test utilizzati per porre diagnosi di diabete mellito o evidenziare stati di rischio per svilupparlo. Deve essere eseguito in pazienti con:
|
| Meccanismo d’azione | L'assunzione del carico di glucosio determina una secrezione insulinica che metabolizza, secondo le condizioni cliniche del soggetto, il glucosio assunto |
| Controindicazioni | Assolute: gastro-duodenoresecati, bendaggio gastrico, by-pass gastroenterici, contemporaneo uso di farmaci iperglicemizzanti Relative: diabete mellito noto, patologie infiammatorie o degenerative del tratto gastroenterico, malattie intercorrenti, allettamento |
| Condizioni preliminari | Ingestione di almeno 150 g/die di carboidrati nei 3 giorni prima dell’esame; da un minimo di 10 ad un massimo di 16 ore di digiuno prima del test; inizio del test tra le 7 e le 9 del mattino |
| Materiale necessario per l’esecuzione | Glucosio anidro da pesare nella quantità di 75 g, poi sciogliere in 300 mL di acqua (o the non zuccherato) |
| Relazione con età, sesso, peso corporeo, gravidanza | In gravidanza esecuzione precoce nelle donne ad elevato rischio di DM Altrimenti è preferibile l'esecuzione tra la 24° e la 28° settimana di gestazione |
| Precauzioni | Nessuna |
| Procedure abbinabili | Sconsigliate |
| Esecuzione | Dopo il prelievo basale, il paziente ingerisce la soluzione di glucosio (in 5-10 minuti), seguita da prelievo a 120’ (tempi: 0, + 120’). Il paziente deve rimanere a riposo, senza assumere alimenti o bevande; non deve fumare |
| Dosaggio | Glicemia (ed eventualmente insulinemia) |
| Possibili effetti collaterali | Nausea, vomito |
| Parametri da monitorare durante l’esecuzione | Nessuno |
| Manovre da eseguire dopo la fine del test | Nessuna |
| Valutazione risultati | Definisce lo stato di normalità se la glicemia plasmatica a digiuno è < 100 mg/dL (5.6 mmol/L) e la glicemia a 2 ore dal carico è ≤ 139 mg/dL (7.7 mmol/L) |
| Interpretazione | Secondo i criteri WHO 2006 e ADA 2013, la diagnosi di diabete può essere stabilita se la glicemia su un campione di plasma raccolto 2 ore dopo il carico di glucosio è ≥ 200 mg/dL (≥ 11.1 mmol/L) (1-2). Il dato dovrebbe essere confermato in un altro giorno con una misurazione ripetuta o dello stesso test oppure confermata da un valore di glicemia a digiuno > 126 mg/dL (7 mmol/L) oppure da HbA1c > 6.5% (48 mmol/mol) (3). Il test consente inoltre di identificare una condizione di rischio di sviluppare diabete mellito (IGT – impaired glucose tolerance) se il valore di glicemia plasmatica dopo 2 ore dal carico è compreso tra 140 e 199 mg/dL (tra 7.7 e 11.0 mmol/L). Approssimativamente il 25% dei soggetti con IGT progredisce verso DM tipo 2 nell’arco di 3-5 anni. Numerosi studi inoltre hanno dimostrato che l’iperglicemia post-prandiale è strettamente associata con il rischio cardiovascolare e la mortalità (4). |
| Attendibilità e ripetibilità dei risultati |
Elevate attendibilità e riproducibilità |
| Giudizio complessivo costo beneficio e costo-efficacia |
Basso costo e elevata efficacia |
| Bibliografia |
|
Test al glucagone nella diagnostica del diabete
Gabriele Zardini
UOC Medicina Generale ad indirizzo Endocrinologico, Azienda Ospedaliera Universitaria Integrata di Verona
| Test al glucagone nella diagnostica del diabete | |
| Indicazioni | Valutazione della riserva di secrezione di insulina (1) nei casi in cui la presenza di anticorpi anti-insulina interferisce con il dosaggio dell’insulinemia, per stabilire il trattamento ipoglicemizzante adeguato e in alcuni casi per differenziare il DM tipo 1 dal tipo 2 (2) |
| Meccanismo d’azione | La somministrazione di glucagone determina iperglicemia, cui segue la rapida secrezione di insulina e C-peptide |
| Controindicazioni | Insufficienza renale; glicemia > 250 mg/dL o C-peptide < 0.2 nmol/L al basale, in quanto l’iperstimolazione della ß-cellula che ne deriva indurrebbe una sovrastima della secrezione insulinica |
| Materiale necessario per l’esecuzione | Glucagone fiale 1 mg |
| Relazione con età, sesso, peso corporeo, gravidanza | Nessuna |
| Precauzioni | Digiuno da almeno 8 ore, senza aver assunto terapia anti-diabetica |
| Esecuzione |
Posizionamento di ago-cannula venosa mantenuta pervia con soluzione "fisiologica"; al tempo -1’ prelievo ematico, preceduto da scarto di 3 mL; al tempo 0, iniezione endovenosa di glucagone, in 1 minuto; al tempo +6’ prelievo, preceduto da uno scarto di 3 mL |
| Dosaggio | Glucosio, C-peptide, insulina. Il dosaggio del C-peptide è preferibile a quello dell’insulina, con la quale è secreto in quantità equimolare, perché non risente della variabilità dell’estrazione epatica, ha una maggiore stabilità, subisce un’influenza trascurabile da parte dell’insulina esogena e quasi nulla da parte degli analoghi |
| Possibili effetti collaterali | Nausea, incremento della pressione arteriosa, di rado sincope (3) |
| Parametri da monitorare durante l’esecuzione | Pressione arteriosa |
| Manovre da eseguire dopo la fine del test | Nessuna |
| Interpretazione | Valori del C-peptide: basale 0.5-3.0 ng/mL; è considerato normale l'aumento dopo stimolo del 150-300% rispetto ai valori basali |
| Attendibilità e ripetibilità dei risultati | Elevate attendibilità e riproducibilità |
| Giudizio complessivo costo beneficio e costo-efficacia | Elevata efficacia nel valutare la riserva insulinica Costo moderato |
| Bibliografia |
|
Clamp gluco-insulinemico
Termine originato dall’inglese “to clamp”, che significa “tenere fermo”, sta ad indicare una metodica in cui si mantiene costante la glicemia a fronte di alcune perturbazioni apportate all’omeostasi del sistema.
Valutazione della sensibilità insulinica
Per sensibilità insulinica si intende l’efficacia dell’insulina nell'esercitare i suoi effetti biologici. In vivo viene comunemente intesa come la capacità da parte dell’ormone di stimolare il metabolismo del glucosio. È compromessa tipicamente nel diabete mellito tipo 2, dove svolge un ruolo patogenetico importante, ma anche in numerose altre condizioni quali ad esempio l’obesità, la sindrome metabolica, la sindrome dell’ovaio policistico.
La valutazione della sensibilità insulinica viene effettuata in genere attraverso il calcolo di indici surrogati basati sul dosaggio di glicemia e insulinemia, a digiuno e dopo carico orale di glucosio (1,2) oppure attraverso test, quali l’ITT o l’IVGTT, tuttavia la metodica gold standard per la misurazione di questo parametro é rappresentata dal clamp euglicemico iperinsulinemico (3).
Clamp euglicemico-iperinsulinemico
Rappresenta attualmente la metodica più affidabile per valutare in vivo la sensibilità insulinica (4).
Consiste nell’innalzare i livelli insulinemici a un valore prestabilito attraverso un’infusione costante di insulina e nella contemporanea infusione di glucosio per mantenere l’euglicemia. L’infusione di glucosio sarà variabile e aggiustata nel corso del test, sulla base del valore della glicemia controllata frequentemente. La quantità di glucosio infuso è espressione della risposta tissutale del singolo individuo all’insulina.
La metodica prevede di predisporre nel paziente a digiuno un duplice accesso venoso, uno per l’infusione di insulina e glucosio e l’altro per i prelievi di sangue su cui valutare la glicemia ed eventuali altri parametri di interesse. È importante che la glicemia sia mantenuta in un range ristretto (intorno a 90 mg/dL), evitando in particolare l’ipoglicemia per la conseguente risposta contro-regolatoria. Per questo motivo è indispensabile effettuare il controllo della glicemia ogni 5-10 minuti e disporre di un analizzatore del glucosio per avere il dato preciso in tempo reale. Inoltre, per evitare un errore di misura legato all’aumentata captazione di glucosio dovuta alla procedura, la misurazione della glicemia deve essere effettuata su sangue venoso “arterializzato”. Ciò si ottiene riscaldando l’arto su cui si effettuano i prelievi (ad esempio con una scatola termostatata). Il test va protratto per un tempo sufficiente, abitualmente non inferiore a 2 ore, per raggiungere un pieno effetto dell’insulina, ovvero un equilibrio del sistema (“steady-state”). Va tenuto presente che il fegato è in grado di immettere in circolo glucosio che si aggiunge a quello infuso. Pertanto, per avere una misura corretta della sensibilità dei tessuti all’insulina è necessario misurare la produzione epatica di glucosio, attraverso l’uso di traccianti (glucosio marcato con isotopi) o in alternativa utilizzare velocità di infusione di insulina più elevate in modo da sopprimere completamente la produzione endogena di glucosio.
Il clamp euglicemico classico prevede una velocità di infusione insulinica di 40 mU/m2 x minuto (= 1 mU/kg x min) che porta l’insulinemia a valori intorno a 80-100 mU/L. A seconda dell’obiettivo, tale velocità di infusione può essere diversificata e può essere anche effettuato un clamp a più step, utilizzando velocità crescenti di infusione insulinica. Va ricordato che durante l’infusione di insulina va monitorata anche la potassiemia e predisposta un’appropriata infusione di potassio, per il noto effetto ipokaliemizzante dell’ormone.
La misura della sensibilità insulinica fornita dal clamp (definita come M”, che sta per glucosio metabolizzato) è data dalla quantità di glucosio infusa nell’unità di tempo durante lo stato stazionario (in condizioni di iperinsulinemia ed euglicemia stabili) ed è espressa in mg/kg di peso x minuto. Il valore di normalità in una popolazione di giovani adulti sani normopeso è di 5-9 mg/kg x min. Dato che il muscolo è il principale utilizzatore periferico del glucosio sotto stimolo insulinico (5), è preferibile normalizzare i dati per la massa magra (MFFM), valutata ad esempio con bio-impedenzometria. Se le concentrazioni di insulinemia raggiunte nel clamp non sono confrontabili con quelle della popolazione di riferimento, si esprime il dato di sensibilità dividendo M per la concentrazione di insulina raggiunta allo steady-state (M/I, MFFM/I). Se condotto appropriatamente, il clamp è una procedura che fornisce misure ben riproducibili della sensibilità insulinica, con coefficienti di variazione di poco superiori al 5%.
Poiché la metodica è complessa, l’uso di questo test è limitato ad ambiti di ricerca. Va ricordato comunque che è l’unica metodica a fornire una misura quantitativa diretta dell’utilizzazione insulino-stimolata del glucosio. Oltre a permettere all’operatore di definire il livello insulinemico da raggiungere, il clamp può essere combinato con altre tecniche, quali ad esempio l’infusione di isotopi del glucosio e di altri substrati per valutare la produzione epatica di glucosio e per studiare il metabolismo intermedio, e la calorimetria indiretta per quantificare l’ossidazione dei substrati. Rimane tuttavia una metodica invasiva, che richiede personale esperto e comporta un significativo consumo di tempo e risorse. Va anche ricordato che la M misura selettivamente l’azione insulinica sul metabolismo glucidico e che può risentire di variazioni nell’utilizzo del glucosio non insulino-mediato.
Clamp iperglicemico-iperinsulinemico
È una metodica che permette di valutare la secrezione ß-cellulare in risposta al glucosio e di quantificare la quota di glucosio metabolizzata dall’organismo in seguito ad uno stimolo iperglicemico.
Come per il clamp euglicemico, sono necessari 2 accessi venosi: uno per l’infusione di glucosio, l’altro per i prelievi di sangue venoso “arterializzato”. Nel clamp iperglicemico la glicemia viene innalzata e mantenuta a 200 mg/dL (11 mmol/L) attraverso un’infusione di glucosio la cui velocità viene modificata continuamente sulla base del valore di glicemia. L’iperglicemia provoca un aumento progressivo nella risposta secretoria ß-cellulare (in questo test non si raggiunge mai uno steady-state nelle concentrazioni circolanti di insulina).
I livelli di insulina o C-peptide raggiunti costituiscono una misura della secrezione ß-cellulare. L’utilizzazione del glucosio (M) può essere calcolata dalla quantità di glucosio infuso, tenendo conto della perdita urinaria di glucosio (calcolata sulle urine raccolte durante il test) e dall’eventuale quantità di glucosio infusa in eccesso o in difetto (correzione sulla base dello spazio di distribuzione del glucosio)(3).
Il clamp iperglicemico, tuttavia, viene raramente utilizzato con lo scopo di valutare secrezione insulinica e utilizzazione del glucosio, piuttosto viene utilizzato per indagare aspetti di fisiopatologia in condizioni standardizzate di iperglicemia.
Bibliografia
- Muniyappa R, Lee S, Chen H, Quon MJ. Current approaches for assessing insulin sensitivity and resistance in vivo: advantages, limitations, and appropriate usage. Am J Physiol Endocrinol Metab 2008, 294: E15-26.
- Buchanan TA, Watanabe RM, Xiang AH. Limitations in surrogate measures of insulin resistance. J Clin Endocrinol Metab 2010, 95: 4874-6.
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979, 237: E214-23.
- Ferrannini E, Mari A. How to measure insulin sensitivity. J Hypertens 1998, 16: 895-906.
- DeFronzo RA. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes 1988, 37: 667-87.
Anticorpi nella diagnostica del diabete
Barbara Pirali
Ambulatori Endocrinologia e Diabetologia, Humanitas Mater Domini, Castellanza (VA)
(aggiornato al 4 ottobre 2015)
Definizioni ed epidemiologia
La distruzione della ß-cellula pancreatica è responsabile del diabete mellito di tipo 1 (DM-1) o immuno-mediato e del diabete autoimmune latente dell'adulto o LADA. Tale distruzione si accompagna alla comparsa di anticorpi anti-insula pancreatica, che si possono identificare nel siero del paziente anni prima dell'esordio clinico della malattia (1,2).
I primi auto-anticorpi isolati, diretti contro il citoplasma delle cellule insulari (ICA), sono i marcatori più sensibili e specifici nella diagnosi di DM-1, ma, a causa della loro difficile determinazione e standardizzazione, si ricorre nell'uso clinico alla ricerca dei cosiddetti "anticorpi biochimici", anticorpi antigene-specifici diretti contro 3 principali antigeni insulari (3), rappresentati da:
- insulina nativa (anticorpi anti-insulina o IAA);
- decarbossilasi dell'acido glutammico (anticorpi anti-GAD65 o GADA);
- due tirosin-fosfatasi, la IA-2A e la IA-2b (anticorpi IA-2A e IA-2b).
Anticorpi anti-insulari si riscontrano nell'85-90% dei pazienti con DM-1 all'esordio clinico e sono presenti nel 5-30% degli adulti (7% nella popolazione italiana) con diabete mellito esordito fenotipicamente come tipo 2, ma che evolve rapidamente verso l'insulino-dipendenza (LADA). Circa il 20% dei soggetti diabetici esprime alla diagnosi un solo auto-anticorpo positivo. Nel LADA prevalgono i GAD65A (4).
Ricerca degli anticorpi: a chi e quando
Il dosaggio degli anticorpi anti-insulari non è raccomandato nella diagnosi di routine del diabete mellito (5). Può però consentire una precisa classificazione eziologica del diabete e soprattutto l'identificazione precoce di quella sottopopolazione di diabetici tipo 2 affetta da LADA, nei quali l'uso precoce di insulina può preservare la funzione ß-cellulare e garantire l’ottimizzazione metabolica (4,5).
In età pediatrica: in bambini non obesi (in condizioni di benessere e in assenza di farmaci iperglicemizzanti) con glicemia a digiuno >100 mg/dL, riconfermata, è opportuno ricercare la presenza di auto-anticorpi contro le ß-cellule (GADA, IA2, anti-insulina), in quanto una positività di queste indagini risulta indicativa di una condizione di rischio per DM-1 e richiederà un attento follow-up per evitare che si ponga diagnosi tardiva di DM-1 con possibile comparsa di cheto-acidosi (5).
| Categorie in cui ricercare gli auto-anticorpi |
| Identificazione di soggetti che richiedono un precoce trattamento insulinico (LADA) Ausilio nella classificazione del diabete Identificazione di soggetti ad aumentato rischio di sviluppare DM-1 Studio della storia naturale del diabete Valutazione dell'efficacia degli studi di intervento |
Il rischio di sviluppare diabete nei familiari di 1° grado dei pazienti con DM-1 è del 5% (15 volte superiore a quello della popolazione generale). La presenza di anticorpi insulari conferisce a questi soggetti un rischio più elevato di sviluppare la malattia, rischio che aumenta con il numero di tipologie di anticorpi presenti. Lo screening sistematico dei parenti di pazienti con DM-1 non è però raccomandato al di fuori dei protocolli di ricerca. La determinazione degli anticorpi anti-insulari è raccomandata per lo screening dei familiari non diabetici che desiderano donare una parte del loro pancreas a un parente con DM-1 terminale. (6)
| Valore diagnostico dei diversi anticorpi | |
| Anticorpo | Significato clinico |
| Ab anti-GAD (GADA) | Presenti nel 70-80% dei soggetti con DM-1 all'esordio clinico Meno frequenti nei bambini che sviluppano DM-1 prima dei 10 anni Rappresentano i marcatori più sensibili per la diagnosi di LADA (talora gli unici identificabili) Prevalgono (a titolo più elevato) nei diabetici con altre patologie auto-immunitarie associate (es. tiroidite) Sono il marcatore più sensibile per definire una positività multipla ad anticorpi anti-insulari |
| Anticorpi anti-insulina (IAA) | Più caratteristici del diabete infantile, con maggiore sensibilità diagnostica (50-60%) nei < 10 anni, mentre dopo i 10 anni la sensibilità diagnostica è < 10% Non hanno utilità clinica nella diagnosi di LADA (presenti in < 1% dei soggetti) |
| Ab IA-2 | Presenti nel 32-75% dei soggetti con DM-1 all'esordio clinico (gli IA-2b nel 20%) La loro frequenza si riduce all'aumentare dell'età di esordio del DM-1 Hanno scarso valore per la diagnosi di LADA (in quanto la loro presenza è quasi sempre associata a quella dei GADA) Sono altamente predittivi di futura comparsa della malattia in parenti di 1° grado di soggetti con DM-1 |
| ICA (Islet cell antibodies) | Presenti in oltre il 90% dei pazienti con DM-1 all'esordio clinico Il 50% dei parenti di 1° grado di pazienti con DM-1 in cui vengono identificati gli ICA, sviluppa diabete entro 9 anni (il loro valore predittivo aumenta al 63% se coesiste positività per IAA) Rappresentano a tutt'oggi il singolo test con la maggior sensibilità diagnostica |
Nelle categorie di pazienti che devono essere sottoposti a screening è stato proposto un pannello anticorpale che prevede:
- nello screening iniziale il dosaggio di GADA e IA-2A (aggiungendo il dosaggio di IAA nei soggetti più giovani);
- qualora un solo marcatore risulti positivo, dovrebbero essere determinati gli ICA (6).
Il monitoraggio degli auto-anticorpi insulari nel corso del follow-up non è clinicamente utile (5,6).
Bibliografia
- Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspective on disease pathogenesis and treatment. Lancet 2001, 358: 221-9.
- Pihoker C, et al. Autoantibodies in diabetes. Diabetes 2005, 54 suppl 2: S52-61.
- Verge CF, et al. Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: Combinatorial Islet Autoantibody Workshop.Diabetes 1998, 47: 1857-66.
- Genovese S, et al. Clinical phenotype and beta-cell autoimmunity in Italian patients with adult onset diabetes. Eur J Endocrinol 2006, 154: 441-7.
- AMD-SID. Standard Italiani per la cura del diabete mellito. 2014.
- ADA. Diagnosis and classification of diabetes mellitus. Diabetes Care 2007, 30 suppl 1: 42-7.