Gastrinomi

Antongiulio Faggiano, Valeria Ramundo, Michela Del Prete, Francesca Marciello, Vincenzo Marotta
Dipartimento di Medicina e Chirurgia Clinica, Università Federico II, Napoli
Definizione, epidemiologia e clinica
Il gastrinoma è un GEP-NET funzionante, solitamente localizzato nel duodeno o nel pancreas, che secerne gastrina e provoca la sindrome di Zollinger Ellison (ZES). La ZES è caratterizzata da ipersecrezione acida, che porta a grave malattia peptica ulcerosa e malattia da reflusso gastroesofageo (GERD)(1-4).
L’incidenza di gastrinoma è di 0.5-2 casi/milione/anno. Il gastrinoma è uno dei più comuni GEP-NET funzionanti nella popolazione generale (5) e compare nel 25-40% dei pazienti affetti da MEN-1 (6,7). In questi, compare a un’età più precoce (in media 32-35 anni) che nei casi sporadici (3,6.7).
Il gastrinoma pancreatico può svilupparsi in qualunque porzione della ghiandola, mentre quello duodenale colpisce più frequentemente la prima parte del duodeno, bulbo compreso (8,9). Alla chirurgia il 70-85% dei gastrinomi viene reperito nel cosiddetto “triangolo del gastrinoma” nel quadrante superiore destro, in corrispondenza del duodeno e della testa del pancreas (8-10).
I principali sintomi classicamente associati a ZES, dovuti a ipersecrezione acida gastrica, sono dolore addominale (75-98% dei casi), diarrea (30-73%), pirosi (44-56%), sanguinamento gastroenterico (44-75%), nausea e vomito (12-30%), calo ponderale (7-53%)(3,4,11).
All’esordio clinico, oltre il 97% dei pazienti ha ipergastrinemia, l’87-90% ha marcata ipersecrezione gastrica acida (> 15 mEq/h) e il 100% pH gastrico < 2 (1,12).
Il tasso di malignità è alto e il 30-40% dei pazienti affetti ha metastasi epatiche (13).
La maggioranza dei pazienti si presenta con una singola ulcera duodenale, sintomi ulcerosi e da GERD, complicanze di ulcera e diarrea. È meno frequente oggigiorno vedere pazienti con ulcere multiple o in localizzazioni insolite (1,3-6,12,14-16). I sintomi possono essere in parte mascherati dall’ampio uso odierno di farmaci anti-secretori, come gli inibitori di pompa e gli anti-H2. La diagnosi deve essere ipotizzata sulla base di una lunga storia ulcerosa o di GERD o della loro recidiva dopo trattamento (6,12,15-17). Questo ritardo può portare alla diagnosi di gastrinoma ad uno stadio più avanzato. La malattia va quindi sempre sospettata in caso di:
- malattia ulcerosa recidivante, grave o familiare;
- malattia ulcerosa:
- senza Helicobacter pylori o altri fattori di rischio;
- associata a grave GERD;
- resistente al trattamento o associata a complicanze (penetrazione, perforazione, sanguinamento);
- associata a endocrinopatie o diarrea (che si risolve con gli inibitori di pompa);
- con pliche gastriche prominenti all’EGDS;
- MEN-1.
Diagnostica
Le prime tappe nella diagnosi di ZES sono anamnesi ed esame obiettivo. Bisogna escludere fattori confondenti, come l’uso di ASA e FANS (18).
In tutti i pazienti con ZES bisogna sempre pensare alla MEN-1, specialmente con anamnesi personale o familiare positiva per endocrinopatie, urolitiasi, altri NET (6,19). Vista l’alta penetranza dell’iperparatiroidismo primario nella MEN-1 (20), la prima tappa di screening è il dosaggio di calcemia e PTH.
Per quanto riguarda la diagnostica di laboratorio, la gastrinemia a digiuno è un eccellente test di screening, con sensibilità > 98%. Bisogna escludere accuratamente i possibili falsi positivi.
La diagnosi di ZES richiede livelli inappropriatamente alti di gastrinemia associati ad aumento della secrezione acida gastrica (> 15 mEq/h o > 5 mEq/h nei gastrectomizzati) e pH gastrico 1000 pg/mL fa la diagnosi, mentre un pH gastrico > 2 la esclude (6). Il problema sono i farmaci interferenti in un paziente pesantemente sintomatico: nei soggetti in terapia cronica gli inibitori di pompa andrebbero sospesi per almeno una settimana (il tempo ottimale teorico sarebbe di 4 settimane)(6,21). Nei pazienti che con la sospensione sarebbero a rischio di sanguinamento, disidratazione e ipopotassiemia da diarrea, visto che l’effetto degli anti-H2 sula secrezione acida è meno pronunciato (22,23), si possono sostituire gli inibitori di pompa con gli anti-H2 per almeno una settimana, sotto stretta supervisione (24,25).
Nei casi dubbi si può eseguire un test con la secretina (19,26-29), che richiede comunque un wash-out farmacologico, o un test di stimolo con calcio, con minore accuratezza diagnostica e maggiori effetti collaterali (19). Non devono più essere eseguiti i test di stimolo della secrezione acida gastrica (2).
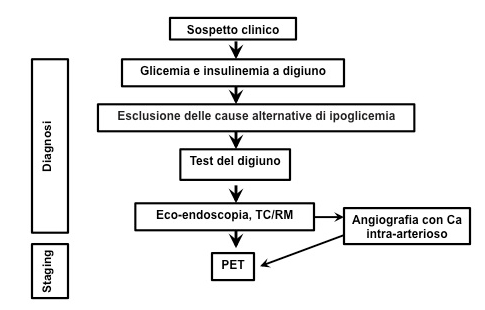
Dopo aver fatto la diagnosi biochimica, si deve eseguire la EGDS. Nella ZES si evidenzia malattia ulcerosa distalmente al bulbo duodenale, nella parte discendente del duodeno o anche più distalmente fin nel digiuno. Le ulcere possono essere multiple a indicare evidente ipersecrezione acida (6).
Si deve poi passare alla localizzazione del tumore e alla valutazione dell’estensione di malattia, tramite TC e/o RM con contrasto, eco-endoscopia, Octreoscan o PET/TC con Gallio, angiografia con iniezione selettiva intra-arteriosa di calcio (6,19). Le stesse metodiche potranno essere usate per valutare la risposta ai trattamenti. È da notare che un’accurata localizzazione del tumore può consentire resezione chirurgica completa, diminuzione della metastatizzazione e aumento della sopravvivenza (19,30-32).

Flow-chart diagnostica per il sospetto gastrinoma
Terapia
I principali approcci terapeutici sono chirurgico e farmacologico.
Attualmente c’è consenso sull’indicazione a una chirurgia radicale curativa nei pazienti con gastrinoma sporadico potenzialmente resecabile e senza gravi controindicazioni (19). L’approccio chirurgico è l’unico con la potenzialità di guarire il paziente e deve essere proposto in prima linea ogni volta che sia possibile. Però, nei casi con malattia localmente avanzata o in presenza di tumori duodeno-pancreatici multipli (MEN-1), non è generalmente possibile la radicalità chirurgica, a meno di non adottare approcci molto aggressivi, come la duodeno-cefalo-pancreasectomia o la pancreasectomia totale, gravati da alta morbilità e mortalità e che quindi devono essere eseguiti solo in centri con grande esperienza nella chirurgia pancreatica. Non c’è più indicazione alla gastrectomia totale per il controllo sintomatologico, se non nei rari pazienti (< 1-2%) che non assumono o non tollerano la terapia anti-secretoria orale (19).
La terapia farmacologica comprende il trattamento sintomatico e i farmaci anti-tumorali. Il trattamento sintomatico mira a controllare l’ipersecrezione gastrica acida e a prevenire le complicanze ulcerose e si basa su inibitori di pompa (di prima scelta per la potenza e la lunga durata d’azione) e anti-H2 (19).
Per quanto riguarda la terapia anti-tumorale, gli analoghi della somatostatina sono considerati di prima scelta nel controllo dei sintomi dei NET iperfunzionanti, che viene ottenuto nella maggioranza dei casi. Nei gastrinomi gli analoghi long-acting sono anche in grado di controllare l’ipersecrezione acida attraverso l’inibizione della secrezione della gastrina, anche se le linee guida continuano a indicare gli inibitori di pompa per il controllo sintomatologico (19). Più di recente gli analoghi sono stati proposti come terapia anti-proliferativa di prima linea nei NET pancreatici non operabili, localmente avanzati o metastatici (33). Anche la target therapy è adesso validata nei pazienti con gastrinomi pancreatici, ove a malattia sia inoperabile o in caso di persistenza/recidiva post-chirurgica: sia everolimus, inibitore di mTOR (34), che sunitinib, inibitore di tirosin-chinasi (35), hanno prolungato la sopravvivenza libera da malattia rispetto al placebo in questa categoria di pazienti.
Altre possibili strategie terapeutiche da prendere in considerazione nei casi di malattia inoperabile in progressione sono la terapia radio-recettoriale e il trattamento loco-regionale delle metastasi epatiche, anche se non sono ancora state validate in studi randomizzati.
Bibliografia
- Roy P, Venzon DJ, Shojamanesh H, et al. Zollinger-Ellison syndrome: clinical presentation in 261 patients. Medicine (Baltimore) 2000, 79: 379–411.
- Roy P, Venzon DJ, Feigenbaum KM, et al. Gastric secretion in Zollinger-Ellison syndrome. Correlation with clinical expression, tumor extent and role in diagnosis--a prospective NIH study of 235 patients and a review of 984 cases in the literature. Medicine (Baltimore) 2001, 80: 189–222.
- Jensen RT, Niederle B, Mitry E, et al; Frascati Consensus Conference; European Neuroendocrine Tumor Society. Gastrinoma (duodenal and pancreatic). Neuroendocrinology 2006, 84: 173–82.
- Ellison EC, Johnson JA. The Zollinger-Ellison syndrome: a comprehensive review of historical, scientific, and clinical considerations. Curr Probl Surg 2009, 46: 13–106.
- Faggiano A, Ferolla P, Grimaldi F, et al. Natural history of gastro-entero-pancreatic and thoracic neuroendocrine tumors. Data from a large prospective and retrospective Italian epidemiological study: the NET management study. J Endocrinol Invest 2012, 35: 817-23.
- Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. A prospective study of 107 cases and comparison with 1,009 patients from the literature. Medicine (Baltimore) 2004, 83: 43–83.
- Ramundo V, Milone F, Severino R, et al. Clinical and prognostic implications of the genetic diagnosis of hereditary NET syndromes in asymptomatic patients. Horm Metab Res 2011, 43: 794-800.
- Gibril F, Jensen RT. Advances in evaluation and management of gastrinoma in patients with Zollinger-Ellison syndrome. Curr Gastroenterol Rep 2005, 7: 114–21.
- Kloppel G, Anlauf M. Gastrinoma – morphological aspects. Wien Klin Wochenschr 2007, 119: 579–84.
- Stabile BE, Morrow DJ, Passaro E Jr. The gastrinoma triangle: operative implications. Am J Surg 1984, 147: 25–31.
- Banasch M, Schmitz F. Diagnosis and treatment of gastrinoma in the era of proton pump inhibitors. Wien Klin Wochenschr 2007, 119: 573–8.
- Vinik AI, Woltering EA, Warner RR, et al; North American Neuroendocrine Tumor Society (NANETS). NANETS consensus guidelines for the diagnosis of neuroendocrine tumor. Pancreas 2010, 39: 713–34.
- Jensen RT. Zollinger-Ellison syndrome. In: Doherty GM, Skogseid B, eds. Surgical Endocrinology: Clinical Syndromes. Philadelphia: Lippincott Williams & Wilkins, 2001: 291–344.
- Berna MJ, Hoffmann KM, Serrano J, Gibril F, Jensen RT. Serum gastrin in Zollinger-Ellison syndrome. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2,229 cases from the literature. Medicine (Baltimore) 2006, 85: 295–330.
- Corleto VD, Annibale B, Gibril F, et al. Does the widespread use of proton pump inhibitors mask, complicate and/or delay the diagnosis of Zollinger-Ellison syndrome? Aliment Pharmacol Ther 2001, 15: 1555–61.
- Kulke MH, Anthony LB, Bushnell DL, et al; North American Neuroendocrine Tumor Society (NANETS).NANETS Treatment Guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas 2010, 39: 735–52.
- Arnold R. Diagnosis and differential diagnosis of hypergastrinemia. Wien Klin Wochenschr 2007, 119: 564–9.
- Lau JY, Sung J, Hill C, et al. Systematic review of the epidemiology of complicated peptic ulcer disease: incidence, recurrence, risk factors and mortality. Digestion 2011, 84: 102-13.
- Jensen RT, Cadiot G, Brandi ML, et al; Barcelona Consensus Conference participants. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 2012, 95: 98-119.
- Thakker RV, Newey PJ, Walls GV, et al; Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012, 97: 2990-3011.
- Jensen RT. Consequences of long-term proton pump blockade: highlighting insights from studies of patients with gastrinomas. Basic Clin Pharmacol Toxicol 2006, 98: 4–19.
- Agréus L, Storskrubb T, Aro P, et al. Clinical use of proton-pump inhibitors but not H2-blockers or antacid/alginates raises the serum levels of amidated gastrin-17, pepsinogen I and pepsinogen II in a random adult population. Scand J Gastroenterol 2009, 44: 564-70.
- Kim BW, Lee BI, Kim HK, et al. Influence of long-term gastric acid suppression therapy on the expression of serum gastrin, chromogranin A, and ghrelin. Korean J Gastroenterol 2009, 53: 84-9.
- Campana D, Piscitelli L, Mazzotta E, et al. Zollinger-Ellison syndrome. Diagnosis and therapy. Minerva Med 2005, 96: 187-206.
- Poitras P, Gingras MH, Rehfeld JF. The Zollinger-Ellison syndrome: dangers and consequences of interrupting antisecretory treatment. Clin Gastroenterol Hepatol 2012, 10: 199–202.
- O’Toole D, Grossman A, Gross D, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: biochemical markers. Neuroendocrinology 2009, 90: 194–202.
- Poitras P, Gingras MH, Rehfeld JF. Secretin stimulation test for gastrin release in Zollinger-Ellison syndrome: to do or not to do? Pancreas 2013, 42: 903-4.
- Desir B, Poltras P. Oral pantoprazole for acid suppression in the treatment of patients with Zollinger-Ellison syndrome. Can J Gastroenterol 2001, 15: 795-8.
- Berna MJ, Hoffmann KM, Long SH, et al. Serum gastrin in Zollinger-Ellison syndrome. II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. Evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoral features. Medicine (Baltimore) 2006, 85: 331–64.
- Fraker DL, Norton JA, Alexander HR, et al. Surgery in Zollinger-Ellison syndrome alters the natural history of gastrinoma. Ann Surg 1994, 220: 320–30.
- Norton JA, Fraker DL, Alexander HR, et al. Surgery increases survival in patients with gastrinoma. Ann Surg 2006, 244: 410–9.
- Morrow EH, Norton JA. Surgical management of Zollinger-Ellison syndrome; state of the art. Surg Clin North Am 2009, 89: 1091–103.
- Jann H, Denecke T, Koch M, et al. Impact of Octreotide LAR on tumour growth control as first-line treatment in neuroendocrine tumours of pancreatic origin. Neuroendocrinology 2013, [Epub ahead of print] DOI: 10.1159/000353785.
- Yao JC, Shah MH, Ito T, et al; RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 514-23.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 501-13.
Insulinomi
Antonella Paoloni, Francesca Rota, Valerio Adinolfi, Laura Rizza, Agnese Barnabei, Marialuisa Appetecchia, Roberto Baldelli
UOSD Endocrinologia, Istituto Nazionale Tumori Regina Elena – IRCCS, Roma
DEFINIZIONE ED EPIDEMIOLOGIA
L’insulinoma è un NET che nasce dalle cellule insulino-secernenti delle insule pancreatiche (1,2). Oltre all’insulina, può dare ipersecrezione di altri ormoni e metaboliti (gastrina, ACTH, glucagone, hCG, somatostatina e acido 5-OH-indolacetico). In casi rari la sindrome dipende da iperplasia diffusa delle ß-cellule senza che vi siano tumori identificabili.
L’insulinoma rappresenta la forma più di frequente di NET del pancreas. Ha un’incidenza di 1-3 casi per milione di popolazione/anno ed è maligno in meno del 10% dei casi.
Si riscontra prevalentemente nelle donne rispetto agli uomini ed è più frequente intorno ai 50 anni di età.
Nel 10% dei casi si tratta di forme multiple e nel 5% dei casi è associato alla sindrome MEN-1 (ma la percentuale sale al 50% in caso di tumori multipli).
CLINICA
Il quadro clinico dipende dall’ipoglicemia ma può essere estremamente aspecifico e variabile, per cui spesso passa diverso tempo prima di riuscire a porre diagnosi. Caratteristicamente l'ipoglicemia secondaria a insulinoma si verifica durante il digiuno e i sintomi possono verificarsi per differenti livelli glicemici (solitamente < 55-60 mg/dL).
I sintomi sono insidiosi, ma di solito predominano quelli neuroglicopenici (tab 1), che possono mimare un'ampia varietà di disturbi neurologici e psichiatrici (3-7). È frequente che la presenza di stato confusionale o comportamenti bizzarri sia descritta con precisione dai conviventi piuttosto che dal paziente. I disturbi a carico del SNC possono progredire sino a perdita di coscienza, convulsioni e coma.
| Tabella 1 Sintomi neuroglicopenici dell’ipoglicemia |
|
| Neurologici | Sonnolenza Disturbi visivi (accomodazione, contrazione pupillare, ecc) Irritabilità Confusione Amnesia Parestesie Sindrome convulsiva |
| Psichiatrici | Comportamento inadeguato e bizzarro Deliri Allucinazioni |
Specialmente nella fase iniziale della malattia possono coesistere sintomi e segni adrenergici, dovuti a eccesso di catecolamine (ansia, palpitazioni, tachicardia, astenia profonda, cefalea, tremori, sudorazione fredda e profusa, pallore), ma una descrizione dettagliata di questi si associa più frequentemente a ipoglicemia funzionale che non a insulinoma.
Questi pazienti imparano a convivere con la malattia, a dominare i sintomi e si abituano a un livello di glicemia molto al di sotto dei valori inferiori della norma senza mostrare sintomi.
Il sospetto clinico
L’ipoglicemia non rappresenta un problema frequente nell’adulto non diabetico. La presenza di sintomi rafforza l’importanza clinica del problema, perché in alcuni soggetti normali il digiuno prolungato può provocare ipoglicemia asintomatica. L’ipoglicemia può dipendere da molte cause oltre all’insulinoma (tab 2) (8,9).
| Tabella 2 Diagnosi differenziale delle cause di ipoglicemia |
|
| Farmaci | Insulina, anti-diabetici orali Chinino, pentamidina, indometacina, litio Più raramente: ACE-inibitori, levofloxacina, trimetoprim-sulfametossazolo, eparina |
| Eccessivo introito alcolico | Blocco della liberazione dei depositi di glucosio |
| Insufficienza epatica, renale o cardiaca | Deplezione di substrati per la gluconeogenesi |
| Digiuno di lunga data (anoressia nervosa) | Deplezione di substrati per la gluconeogenesi |
| Tumori non insulari | Eccessiva produzione di IGF-II che consuma il glucosio |
|
Chirurgia gastrica (post bypass gastrico) |
Accelerato transito e malassorbimento |
| Deficit di ormoni contro-regolatori | |
| Ipoglicemia autoimmune |
|
Il sospetto di insulinoma è forte in presenza della triade di Whipple, presente nel 75% dei casi, che comprende (10):
- sintomi di ipoglicemia;
- rilievo di ipoglicemia in corrispondenza dei sintomi;
- regressione dei sintomi con la somministrazione di glucosio.
I sintomi compaiono più frequentemente di notte o nel primo mattino e comunque durante il digiuno, anche se la comparsa di un’ipoglicemia post-prandiale non esclude la diagnosi (11,12). I sintomi possono essere peggiorati da esercizio, ingestione di alcol, dieta ipocalorica e uso di alcuni farmaci (1,2).
L’attivazione da parte dell’ipoglicemia di un riflesso vagale, che comporta la stimolazione della secrezione acida gastrica e della peristalsi e che aumenta la velocità di svuotamento gastrico, porta ad aumento dell’appetito. Il paziente affetto da insulinoma impara a mangiare ogni 2-3 ore e nel 20-40% dei casi cresce di peso (fino all’obesità).
DIAGNOSI
Il primo passo è la conferma dell'ipoglicemia con livelli inappropriati di insulina. Sono diagnostici livelli documentati di insulinemia > 3 μU/mL (18 pmol/L), in assenza di metaboliti delle sulfoniluree nel plasma o nelle urine.
Oltre al dosaggio dell’insulina, è importante la valutazione dei livelli di peptide C, che vengono considerati diagnostici se > 0.6 ng/mL (0.2 nmol/L), e, se disponibili, di proinsulina (> 5.0 pmol/L) (3,4,6,7,9,13). Nei pazienti affetti, la proinsulina arriva al 70% dell’immunoreattività dell’insulina, mentre nel soggetto normale è < 20%.
La diagnosi differenziale deve considerare anche la presenza di possibili ipoglicemie factitie:
- quelle da auto-somministrazione di insulina sono caratterizzate dalla presenza di ipoglicemia, iperinsulinemia, ridotti livelli plasmatici di peptide C e proinsulina e talvolta anche dalla presenza di anticorpi circolanti anti-insulina;
- in quelle conseguenti ad assunzione di sulfaniluree (in cui il peptide C non è basso) è necessario dimostrare la presenza del farmaco o dei suoi metaboliti nel plasma e nelle urine.
Un altro elemento importante per discriminare l’ipoglicemia iperinsulinemica da altre cause, è l’assenza di chetonuria.
Prove funzionali
La diagnosi biochimica si basa sull’incapacità dell’ipoglicemia di sopprimere la secrezione endogena di insulina (14) ovvero sui livelli di insulina inappropriatamente alti per la glicemia. Nel 95% dei casi la diagnosi si ottiene solo durante il test del digiuno protratto, che costituisce il test diagnostico (15).
In passato venivano utilizzati altri test come:
- la prova di tolleranza insulinica, in grado di valutare la sopprimibilità dei livelli plasmatici di peptide C dopo somministrazione di insulina ev (0.1 UI/kg);
- la prova al diazossido (600 mg di diazossido, farmaco con potente azione iperglicemizzante, in 250 cc di soluzione fisiologica, con dosaggio di glicemia e insulinemia ogni 15 minuti per 3 ore), il cui scopo è valutare la soppressione dei valori di insulina;
- la prova da carico orale di glucosio, con prelievi prolungati fino a 3 ore, utilizzata solo per la diagnosi differenziale con le ipoglicemie reattive;
- la prova di soppressione con octreotide (potente inibitore della secrezione di insulina e glucagone), che consiste nella somministrazione in bolo di 125 μg di octreotide seguiti da infusione costante di 250 μg/ora per 180 min.
Diagnostica per immagini
Permette una precisa localizzazione pre-operatoria del tumore e deve essere avviata dopo aver posto la diagnosi biochimica (16). Nella maggior parte dei casi è necessaria l’associazione di più metodiche.
Poiché l’80% degli insulinomi è < 2 cm, l’ecografia trans-addominale ha sensibilità < 50%. L’eco-endoscopia è positiva nel 70-95% dei casi, se eseguita da un endoscopista esperto (17). TC elicoidale e multislice e RM hanno sensibilità sovrapponibile (82-94%)(18,19).
L’Octreoscan è positivo solo nel 50% dei casi di insulinoma localizzato, a causa delle piccole dimensioni e della bassa densità o della mancanza di recettori per la somatostatina che legano l’octreotide con alta affinità (SSTR2)(20).
Risultati promettenti sono stati ottenuti con l’utilizzo di varie metodiche sperimentali PET/TC che non utilizzano i recettori della somatostatina: DOPA-PET e 111In-DOTA-exendin-4 (21, 22).
Angiografia selettiva e stimolazione intra-arteriosa con calcio gluconato
Essendo tumori molto vascolarizzati, l’arteriografia con calcio gluconato (dal momento che il calcio è in grado di stimolare il rilascio di insulina dal tessuto neoplastico e non dal tessuto normale) mediante cateterizzazione selettiva dei rami minori delle arterie gastro-duodenale, mesenterica superiore e splenica, per la ricerca di un gradiente di concentrazione, è positiva nell’88-100% dei casi, con sensibilità dell’82% e specificità del 95% (23-25). Il test è comunque poco disponibile, molto indaginoso e costoso, per cui deve essere riservato solo ai casi con diagnosi biochimica certa e negatività degli altri esami di diagnostica per immagini.
Nonostante l’esecuzione di tutte le tecniche di localizzazione sovradescritte, solo nel 60-70% dei casi si riesce a localizzare il tumore prima dell’intervento. Tra gli altri casi, i pazienti con sintomi ben controllabili con la terapia farmacologica possono essere tenuti in sorveglianza stretta, mentre i casi più gravemente sintomatici devono essere comunque avviati alla chirurgia: l’esplorazione pancreatica da parte di un chirurgo esperto e l’uso dell’ecografia intra-operatoria portano all’identificazione di un tumore in oltre il 90% dei casi (17,26).
Flow-chart diagnostica nel sospetto insulinoma
TERAPIA
Terapia farmacologica
Da utilizzare allo scopo di prevenire le ipoglicemie in pazienti selezionati prima dell’intervento chirurgico, in quelli con alto rischio operatorio e/o dove la terapia chirurgica fallisce o nelle forme maligne non resecabili (27). Esistono diversi approcci terapeutici.
Il diazossido (Proglicem, cp 25-100 mg) è in grado di ridurre l’ipersecrezione di insulina. La dose iniziale è 3 mg/kg in 2-3 dosi refratte ogni 8-12 h; il dosaggio può poi essere aggiustato in base alle necessità fino a 8 mg/kg/die. Deve essere associato a un diuretico tiazidico per controllare gli effetti avversi (edemi e iperpotassiemia)(27).
Gli analoghi della somatostatina long-acting hanno un’efficacia variabile e devono essere presi in considerazione nel trattamento dei pazienti con insulinoma sintomatico e con ipoglicemia continua, che sono refrattari al trattamento con diazossido (28).
Everolimus sembra efficace nei pazienti con insulinoma metastatico e ipoglicemia refrattaria, ma la tollerabilità è da monitorare attentamente (29,30).
Glucagone e cortisonici possono essere utili transitoriamente in condizioni di emergenza.
Nei pazienti metastatici si può utilizzare la chemioterapia (streptozotocina, tossica per le ß-cellule, 1 g/m2 ev settimanale per 4 settimane). Determina risposta parziale nel 50% e completa nel 20% dei pazienti, che aumenta al 33% se associata a 5-fluorouracile (31). Richiede il monitoraggio della funzionalità renale (proteine urinarie, creatininemia) ed epatica e dell'emocromo (potenziale tossicità ematopoietica) e non migliora la sopravvivenza.
Terapia chirurgica
È il trattamento di scelta poiché la resezione dell’insulinoma ottiene la guarigione nel 90% dei casi (32). È importante la preparazione per ridurre il rischio di ipoglicemia intra-operatoria: diazossido il giorno dell’intervento e infusione di glucosata al 10% ad almeno 100 mL/h, con monitoraggio dei livelli glicemici.
I possibili interventi sono:
- enucleazione e/o enucleoresezione
- pancreasectomia sinistra
- resezioni intermedie pancreatiche
- duodeno-cefalo-pancreasectomia (raro)
- chirurgia citoriduttiva.
Nei pazienti con MEN-1 è opportuno eseguire una pancreasectomia subtotale piuttosto che un’enucleazione, vista l’alta probabilità di tumori multipli (32,33).
Le percentuali globali di guarigione chirurgica si avvicinano al 90%. Un piccolo insulinoma singolo, localizzato in corrispondenza o in prossimità della superficie del pancreas, di solito, può essere enucleato chirurgicamente. Nel caso di un singolo adenoma di grandi dimensioni o localizzato in profondità nel corpo o nella coda del pancreas, nel caso di lesioni multiple del corpo o della coda (o di entrambi) o nel caso in cui non venga trovato alcun insulinoma (circostanza insolita), si esegue una resezione pancreatica subtotale distale. In < 1% dei casi, l'insulinoma è localizzato in una sede ectopica, nel tessuto peri-pancreatico della parete duodenale o nell'area peri-duodenale e può essere trovato soltanto con una diligente ricerca. La pancreatico-duodenectomia (intervento di Whipple) viene eseguita nei casi di insulinoma maligno resecabile del pancreas prossimale. La pancreasectomia totale viene eseguita se una precedente resezione pancreatica subtotale non si è dimostrata adeguata.
Nei pazienti con metastasi epatiche non resecabili si può prendere in considerazione la chemioterapia intra-arteriosa o l’embolizzazione dell’arteria epatica (34-37).
PROGNOSI
La maggior parte degli insulinomi sono benigni con risoluzione completa dopo resezione radicale. Le recidive compaiono nel 5% dei casi. Nelle forme maligne metastatiche la sopravvivenza è di 16-26 mesi.
BIBLIOGRAFIA
- Phan GQ, Yeo CJ, Hruban RH, et al. Surgical experience with pancreatic and peripancreatic neuroendocrine tumors: review of 125 patients. J Gastrointest Surg 1998, 2: 473-82.
- Mathur A, Gorden P, Libutti SK. Insulinoma. Surg Clin North Am 2009, 89: 1105-21.
- Senniappan S, Shanti B, James C, Hussain K. Hyperinsulinaemic hypoglycaemia: genetic mechanisms, diagnosis and management. J Inherit Metab Dis 2012, 35: 589-601.
- McAulay V, Deary IJ, Frier BM. Symptoms of hypoglycaemia in people with diabetes. Diabet Med 2001, 18: 690-705.
- Cox D, Gonder-Frederick L, McCall A, et al. The effects of glucose fluctuation on cognitive function and QOL: the functional costs of hypoglycaemia and hyperglycaemia among adults with type 1 or type 2 diabetes. Int J Clin Pract 2002, Suppl Jul (129): 20-6.
- Okabayashi T, Shima Y, Sumiyoshi T, et al. Diagnosis and management of insulinoma. World J Gastroenterol 2013, 19: 829-37.
- Cryer PE, Axelrod L, Grossman AB, et al; Endocrine Society. Evaluation and management of adult hypoglycemic disorders: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 2009, 94: 709–28.
- Davda R, Seddon BM. Mechanisms and management of non-islet cell tumour hypoglycaemia in gastrointestinal stromal tumour: case report and a review of published studies. Clin Oncol (R Coll Radiol) 2007, 19: 265-8.
- Smeeks FC. Hypoglycemia. Medscape Dec 2009.
- Waickus CM, de Bustros A, Shakil A. Recognizing factitious hypoglycemia in the family practice setting. J Am Board Fam Pract 1999, 12: 133-6.
- Placzkowski KA, Vella A, Thompson GB, et al. Secular trends in the presentation and management of functioning insulinoma at the Mayo Clinic, 1987-2007. J Clin Endocrinol Metab 2009, 94: 1069–73.
- Toaiari M, Davì MV, Dalle Carbonare L, et al. Presentation, diagnostic features and glucose handling in a monocentric series of insulinoma. J Endocrinol Invest 2013 Apr 18. [Epub ahead of print].
- Dizon AM, Kowalyk S, Hoogwerf BJ. Neuroglycopenic and other symptoms in patients with insulinomas. Am J Med 1999, 106: 307-10.
- Starke A, Saddig C, Kirch B, et al. Islet hyperplasia in adults: challenge to preoperatively diagnose non-insulinoma pancreatogenic hypoglycemia syndrome. World J Surg 2006, 30: 670-9.
- van Bon AC, Benhadi N, Endert E, et al. Evaluation of endocrine tests. D: the prolonged fasting test for insulinoma. Neth J Med 2009, 67: 274-8.
- Baudin E, Caron P, Lombard-Bohas C, et al, on behalf of the Société française d’endocrinologie and the Groupe d’étude des tumeurs endocrines. Malignant insulinoma: Recommendations for characterisation and treatment. Ann Endocrinol in press (doi.org/10.1016/j.ando.2013.07.001).
- McLean A. Endoscopic ultrasound in the detection of pancreatic islet cell tumours. Cancer Imaging 2004, 4: 84-91.
- Liu Y, Song Q, Jin HT, et al. The value of multidetector-row CT in the preoperative detection of pancreatic insulinomas. Radiol Med 2009, 114: 1232-8.
- Anaye A, Mathieu A, Closset J, et al. Successful preoperative localization of a small pancreatic insulinoma by diffusion-weighted MRI. JOP 2009, 10: 528-31.
- Christ E, Wild D, Forrer F, et al. Glucagon-Like Peptide-1 receptor imaging for localization of insulinomas. J Clin Endocrinol Metab 2009, 94: 4398-405.
- Minn H, Kauhanen S, Seppänen M, Nuutila P. 18F-FDOPA: a multiple-target molecule. J Nucl Med 2009, 50: 1915-8.
- Wild D, Christ E, Caplin ME, et al. Glucagon-like peptide-1 versus somatostatin receptor targeting reveals 2 distinct forms of malignant insulinomas. J Nucl Med 2011, 52: 1073-8.
- Salazar R, Wiedenmann B, Rindi G, Ruszniewskj P. ENETS 2011 consensus guidelines for the management of patients with digestive neuroendocrine tumours: an update. Neuroendocrinology 2012, 95: 71-3.
- Wiesli P, Uthoff H, Perren A, et al. Are biochemical markers of neuroendocrine tumors coreleased with insulin following local calcium stimulation in patients with insulinomas? Pancreas 2011, 40: 995-9.
- Guettier JM, Kam A, Chang R, et al. Localization of insulinomas to regions of the pancreas by intraarterial calcium stimulation: the NIH experience. J Clin Endocrinol Metab 2009, 94: 1074–80.
- Proye C, Malvaux P, Pattou F, et al. Noninvasive imaging of insulinomas and gastrinomas with endoscopic ultrasonography and somatostatin receptor scintigraphy. Surgery 1998, 124: 1134-43; discussion 1143-4.
- Diagnosis and management of pancreatic endocrine tumors. In: De Vita V, Lawrence T, Rosenberg S. Cancer. Principles and Practice of Oncology. 8th ed. Lippincott, Williams and Wilkins, Philadelphia, PA, 2008: 1706-15.
- Arnold R, Simon B, Wied M. Treatment of neuroendocrine GEP tumours with somatostatin analogues: a review. Digestion 2000, 62 Suppl 1: 84-91.
- Bernard V, Lombard-Bohas C, Taquet MC, et al. Efficacy of everolimus in patients with metastatic insulinoma and refractory hypoglycemia. Eur J Endocrinol 2013, 168: 665-74.
- Zhan HX, Cong L, Zhao YP, et al. Activated mTOR/P70S6K signaling pathway is involved in insulinoma tumorigenesis. J Surg Oncol 2012, 106: 972-80.
- Rougier P, Mitry E. Chemotherapy in the treatment of neuroendocrine malignant tumors. Digestion 2000, 62 Suppl 1: 73-8.
- Lo CY, Lam KY, Fan ST. Surgical strategy for insulinomas in multiple endocrine neoplasia type I. Am J Surg 1998, 175: 305-7.
- Dexter SP, Martin IG, Leindler L, et al. Laparoscopic enucleation of a solitary pancreatic insulinoma. Surg Endosc 1999, 13: 406-8.
- Moscetti L, Saltarelli R, Giuliani R, et al. Intra-arterial liver chemotherapy and hormone therapy in malignant insulinoma: case report and review of the literature. Tumori 2000, 86: 475-9.
- Ahlman H, Wangberg B, Jansson S, et al. Interventional treatment of gastrointestinal neuroendocrine tumours. Digestion 2000, 62 Suppl 1: 59-68.
- Ruszniewski P, Malka D. Hepatic arterial chemoembolization in the management of advanced digestive endocrine. Digestion 2000, 62 Suppl 1: 79-83.
- Chandra P, Yarandi SS, Khazai N, Jacobs S. Management of intractable hypoglycaemia with Yttrium-90 radioembolization in a patient with malignant insulinoma. Am J Med Sci 2010, 340: 414-7.
Glucagonomi
Maria Vittoria Davì1 & Chiara Martini2
1UOC Medicina Interna D, Policlinico GB Rossi, AOUI Verona
2Clinica Medica 3^, Azienda Ospedaliera-Università di Padova
Epidemiologia
I NET pancreatici secernenti glucagone (glucagonomi) sono estremamente rari, con un'incidenza stimata di 0.01-0.1 nuovi casi/106 abitanti/anno; si presentano più frequentemente come forme sporadiche, ma sono riportati nel 3% dei pazienti affetti da MEN-1.
I glucagonomi rappresentano il 2% dei tumori pancreatici insulari (1) riportati nel registro SEER. In un recente studio epidemiologico italiano rappresentano l'1.5% di tutte le forme sindromiche (2). In quanto patologia rara, sono poche in letteratura le casistiche monocentriche, tutte con un numero ristretto di pazienti (3-5).
Presentazione clinica
Le modificazioni post-traslazionali del precursore del glucagone portano alla formazione di differenti forme molecolari e non sempre le neoplasie insulari secernono forme biologicamente attive. Si dovrebbero definire glucagonomi solo le forme sindromiche. La sindrome associata al glucagonoma è caratterizzata da diabete mellito e manifestazioni muco-cutanee (eritema necrolitico migrante, distrofie ungueali, stomatite, cheilite angolare, glossite atrofica). Il quadro sindromico venne per la prima volta descritto nel 1942 (6) associato a un tumore pancreatico, ma solo nel 1966 fu documentata l'associazione fra iperglucagonemia e manifestazione cutanea (7), successivamente definita con il termine attuale (8, 9).
L'eritema necrolitico migrante è una manifestazione suggestiva ma non specifica di glucagonoma, potendosi trovare associato ad altre patologie (IBD, cirrosi epatica, morbo celiaco). È presente in circa il 70% dei pazienti con glucagonoma (3), talora come manifestazione unica, talora associata al diabete, e può precedere di anni la diagnosi della neoplasia sottostante. La sua eziopatogenesi permane tuttora un fatto speculativo, ma è probabile che sia multi-fattoriale: vengono ipotizzati un effetto diretto del glucagone, con un meccanismo tuttavia non definito, ma anche un deficit di aminoacidi, componenti del complesso vitaminico B, zinco e acidi grassi essenziali (11,12). Sia dal punto di vista clinico che istologico è assimilabile alle manifestazioni cutanee tipiche di altri stati carenziali (deficit di niacina, zinco, ecc). Da questa ipotesi scaturisce il trattamento infusionale con aminoacidi e acidi grassi suggerito da alcuni autori (13). Il quadro inizia come papule eritematose che coinvolgono prevalentemente il perineo, le pieghe inguinali, il tronco, gli arti e la regione peri-orale, con tendenza alla confluenza. Le papule evolvono, nell'arco di 7-14 giorni, in vescicole, con successiva erosione centrale dovuta alla necrosi della porzione più superficiale dell'epidermide, cui istologicamente corrispondono un'infiltrazione linfocitaria peri-vascolare associata a edema e vacuolizzazione dei cheratinociti. La guarigione inizia dalla regione centrale, tanto che le lesioni assumono un aspetto anulare con caratteristica colorazione bronzina. In genere sono intensamente pruriginose e dolorose.
Il diabete mellito, riportato nel 40-70% delle casistiche, in genere precede le manifestazioni cutanee e, almeno in parte, è secondario all'alterato rapporto fra le concentrazioni di insulina e glucagone a livello epatico, che determina un incremento della produzione epatica di glucosio; generalmente non è di difficile controllo.

Eritema necrolitico migrante associato a glucagonoma della coda del pancreas (grazie a Fernando Cirillo)
Altre manifestazioni aspecifiche ma frequenti sono anoressia e calo ponderale (70%), diarrea (30%), così come le manifestazioni trombo-emboliche(TVP agli arti inferiori, embolia polmonare) e l'anemia normocromica normocitica, verosimilmente secondaria alla malattia cronica ma anche a un effetto diretto inibitorio del glucagone sull'eritropoiesi. Sono descritte anche manifestazioni neuropsichiatriche (depressione, atassia, incontinenza, deficit visivi).
Sede e prognosi
Si localizzano prevalentemente nella regione corpo-coda del pancreas ed, essendo una patologia rara e con manifestazioni iniziali sfumate e non specifiche, sono metastatici alla diagnosi (fegato, linfonodi, osso, polmone) nel 50-100% dei casi, a seconda delle casistiche (3,10). La prognosi è in funzione dello stadio, ma in genere, a fronte del potenziale di malignità, sono tumori a lenta crescita in cui il debulking può produrre almeno un miglioramento sintomatologico. Molte delle manifestazioni della sindrome trovano ragione nell'effetto “catabolico” dell'ormone (fig 1), cui può anche contribuire una co-secrezione di polipeptide pancreatico.

Diagnosi
Per la conferma della diagnosi è fondamentale il rinvenimento di un incremento dei livelli di glucagone, che va differenziato da quello presente in altri stati fisiologici o patologici ma in assenza di neoplasia insulare (digiuno, stato settico, ipercortisolismo, insufficienza epatica e renale). In queste altre situazioni è improbabile un incremento > 500 pg/mL. La riduzione dei livelli serici di multipli aminoacidi rappresenta un riscontro di laboratorio caratteristico, anche se poco utilizzabile nella pratica clinica.
Terapia
L'approccio chirurgico rappresenta l'unico trattamento in grado di condurre a guarigione, anche se perseguibile nella minoranza dei casi a causa di una malattia localmente avanzata o frequentemente metastatica. In tali situazioni i trattamenti loco-regionali possono produrre una palliazione del quadro sindromico, obiettivo che può essere perseguito anche con il supporto nutrizionale e con il trattamento farmacologico con analoghi della somatostatina (14,15).
Bibliografia
- Yao JC, Eisner MP, Leary C, et al. Population-based study of islet cell carcinoma. Ann Surg Oncol 2007, 14: 3492-500.
- Faggiano A, Ferolla P, Grimaldi F, et al. Natural history of gastro-entero-pancreatic and thoracic neuroendocrine tumors. Data from a large prospective and retrospective Italian Epidemiological study: The NET management study. J Endocrinol Invest 2012, 35: 817-23.
- Wermers RA, Fatourechi V, Wynne AG, et al. The glucagonoma syndrome. Clinical and pathologic features in 21 patients. Medicine (Baltimore) 1996, 75: 53–63.
- Kindmark H, Sundin A, Granberg D, et al. Endocrine pancreatic tumors with glucagon hypersecretion: a retrospective study of 23 cases during 20 years. Med Oncol 2007, 24: 330-7.
- Eldor R, Glaser B, Fraenkel M. et al. Glucagonoma and the glucagonoma syndrome-cumulative experience with an elusive endocrine tumour. Clin Endocrinol 2011, 74: 593-8.
- Becker WS, Kahn D, Rothman S. Cutaneous manifestations of internal malignant tumors. Arch Dermatol Syphilol 1942, 45: 1069-80.
- McGavran MH, Unger RH, Recant L. A glucagon-secreting alpha-cell carcinoma of the pancreas. N Engl J Med 1966, 274: 1408–13.
- Wilkinson DS. Necrolytic migratory erythema with pancreatic carcinoma. Proc R Soc Med 1971, 64: 1197-8.
- Wilkinson DS. Necrolytic migratory erythema with carcinoma of the pancreas. Trans S John’s Hosp Dermat Soc 1973, 59: 244–50.
- Stacpoole PW. The glucagonoma syndrome: clinical features, diagnosis, and treatment. Endocr Rev 1981, 2: 347–61.
- Van Beek AP, de Haas ER, van Vloten WA et al. The glucagonoma syndrome and necrolytic migratory erythema: a clinical review. Eur J Endocrinol 2004, 151: 531–7.
- Tierney EP, Badger J. Etiology and pathogenesis of necrolytic migratory erythema: review of the literature. Med Gen Med 2004, 6: 4.
- Alexander EK, Robinson M, Staniec M, et al. Peripheral amino acid and fatty acid infusion for the treatment of necrolytic migratory erythema in the glucagonoma syndrome. Clin Endocrinol (Oxf) 2002, 57: 827–31.
- Boden G, Ryan IG, Eisenschmid BL, et al. Treatment of inoperable glucagonoma with the long acting somatostatin analogue SMS 201-995. N Engl J Med 1986, 314: 1686-9.
- Rosenbaum A, Flourie B, Chagnon S, et al. Octreotide (SMS 201–995) in the treatment of metastatic glucagonoma: report of one case and review of the literature. Digestion 1989, 42: 116–20.
Somatostatinomi
Maria Vittoria Davì1 & Chiara Martini2
1UOC Medicina Interna D, Policlinico GB Rossi, AOUI Verona
2Clinica Medica 3^, Azienda Ospedaliera-Università di Padova
Epidemiologia
Il somatostatinoma rappresenta una rara forma di tumore neuroendocrino, la cui incidenza stimata è di 1 nuovo caso/40 milioni di abitanti/anno. L'età media alla diagnosi si aggira attorno ai 50 anni, senza differenza di genere.
Descritto per la prima volta nel 1977 (1,2), può localizzarsi sia a livello pancreatico, in più del 50% dei casi, che duodenale, nella regione peri-ampollare. Sono stati descritti, seppur raramente, anche a livello di vie biliari, digiuno, colon e retto (3).
In più del 90% dei casi si presenta come forma sporadica, ma può insorgere anche nell'ambito di sindromi familiari quali la neurofibromatosi di tipo 1, in cui la localizzazione più frequente è quella duodenale (4), la MEN-1 caratteristica per la presenza di metastasi, unitamente alla localizzazione pancreatica. Più del 70% delle forme pancreatiche è metastatico alla diagnosi.
Presentazione clinica
Rimane motivo di dibattito se definire somatostatinomi solo i NET in cui è manifesta la sindrome o anche quelli in cui vi sia solo la positività immuno-istochimica. Dal punto di vista clinico possono manifestarsi con una sintomatologia aspecifica, particolarmente le forme duodenali (ittero ostruttivo, algie addominali anche legate a quadri subocclusivi), e il loro riscontro può anche essere del tutto incidentale. La cosiddetta “sindrome inibitoria”, ipotizzata per la prima volta nel 1979 (5) e legata all'effetto inibitorio della somatostatina sulla funzione colecistica, sulle differenti secrezioni endocrine, esocrine e sull'assorbimento intestinale, è costituita da diabete mellito, colelitiasi e diarrea/steatorrea (6,7). Possono associarsi ipocloridria, dispepsia, anemia e calo ponderale. Il quadro sindromico è più frequente nelle forme pancreatiche (> 90%), generalmente di maggiori dimensioni e associate a una maggior produzione di somatostatina, rispetto a quelle duodenali (< 20%).
Diagnosi
Oltre che sulla clinica e sulla diagnostica per immagini, si basa sui livelli plasmatici di somatostatina e non vi è un test diagnostico provocativo attendibile di conferma (in passato utilizzata la Tolbutamide ev) (6).
Dal punto di vista istologico, le forme duodenali si caratterizzano per la presenza di corpi psammomatosi (49-68%), raramente descritti nei tumori pancreatici (8,9); l'immuno-istochimica documenta un'intensa immuno-reattività per la somatostatina in una frazione cellulare predominante, anche nei casi in cui non è clinicamente presente la sindrome.
Trattamento
La resezione chirurgica rimane l'unico strumento terapeutico in grado di portare a guarigione le forme non estesamente metastatiche al momento della diagnosi.
Bibliografia
- Ganda OP, Weir GC, Soeldner JS, et al. ‘Somatostatinoma’: a somatostatin-containing tumor of the endocrine pancreas. N Engl J Med 1977, 296: 963–7.
- Larsson LI, Hirsch MA, Holst JJ, et al. Pancreatic somatostatinoma: clinical features and physiological implications. Lancet 1977, 1: 666-8.
- Nesi G, Marcucci T, Rubio CA, et al. Somatostatinoma: Clinico-pathological features of three cases and literature reviewed. J Gastroenterol Hepatol 2008, 23: 521–6.
- Fendrich V, Ramaswamy A, Slater EP, et al. Duodenal somatostatinoma associated with Von Recklinghausen's disease. J Hepatobiliary Pancreat Surg 2004, 11: 417-21.
- Krejs GJ, Orci L, Conlon JM, et al. Somatostatinoma syndrome. N Engl J Med 1979, 301: 285-92.
- Stacpoole PW, Kasselberg AG, Berelowitz M, et al. Somatostatinoma syndrome: does a clinical entity exist? Acta Endocrinol 1983, 102: 80–7.
- Moayedoddin B, Booya F, Wermers RA, et al. Spectrum of malignant somatostatin-producing neuroendocrine tumors. Endocr Pract 2006, 12: 394–400.
- Green BT, Rockey DC. Duodenal somatostatinoma presenting with complete somatostatinoma syndrome. J Clin Gastroenterol 2001, 33: 415–7.
- Tanaka S, Yamasaki S, Matsushita H, et al. Duodenal somatostatinoma: a case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol Int 2000, 50: 146-52.
VIPomi
Maria Vittoria Davì1 & Chiara Martini2
1UOC Medicina Interna D, Policlinico GB Rossi, AOUI Verona
2Clinica Medica 3^, Azienda Ospedaliera-Università di Padova
Epidemiologia
Il Vipoma è un tumore neuroendocrino (NET) raro, con un’incidenza di 0.1 caso/milione di individui/anno (1). La sua prevalenza tra i NET pancreatici è dello 0.6-1% e l’età alla presentazione è tra i 48-51 anni (2-3). Nella maggior parte dei casi è sporadico, ma può presentarsi associato alla MEN-1 nell’ 8.7% dei casi. Nel 4% dei casi può essere multifocale (1).
Sede
Nella maggior parte dei casi è localizzato nel pancreas, prevalentemente alla coda, spesso di grandi dimensioni e metastatico al fegato già al momento della diagnosi. Sedi meno frequenti sono retro-peritoneo, digiuno, mediastino, polmoni e gangli simpatici (1,2).
Presentazione clinica
Il VIP (Vasoactive Intestinal Polipeptide) stimola la secrezione pancreatica e intestinale, inibisce l’assorbimento di elettroliti e acqua nell’intestino e la secrezione gastrica acida; inoltre, determina riassorbimento osseo, glicogenolisi e vasodilatazione.
Questi effetti sono responsabili della presentazione clinica del VIPoma, caratterizzata da diarrea, presente nel 100% dei casi, ipokaliemia, acloridria, perdita di peso (45%), acidosi metabolica, ipercalcemia (50%), intolleranza ai carboidrati (50%) e flushing (0-33%) (2-5). La diarrea è tipicamente secretoria, non recede con il digiuno, è caratterizzata da abbondante volume fecale (> 3 L/die di feci acquose) e non risponde al trattamento anti-diarroico. Può essere intermittente nel 57% o continua nel 47% circa dei casi (1). L’ipokaliemia può essere molto grave (< 2.5 mEq/L), con rischio di aritmie cardiache e morte improvvisa. Le metastasi sono meno frequenti (29%) nei pazienti affetti da Vipomi che originano dai gangli, e coinvolgono soprattutto i linfonodi (2).
Diagnosi
Si basa sulla caratteristica sindrome clinica e sull’aumento dei valori di VIP (4-7). Tale dosaggio ormonale non è diffusamente disponibile (nel Veneto è possibile dosarlo presso il laboratorio dell’Azienda Ospedaliera Universitaria di Padova). La Cromogranina A, un marker generale di NET, non è utile nella diagnosi di VIPoma, ma può avere un ruolo nel monitoraggio della risposta ai trattamenti (8). Inoltre possono essere cosecreti altri ormoni, come Polipeptide Pancreatico, Calcitonina, Gastrina, Glucagone, Insulina, Somatostatina, GHRH (5).
Per quanto riguarda la localizzazione del tumore, viene eseguita mediante TC o RMN addome, completata da ® Octreoscan o, se disponibile, 68Ga-DOTATOC-PET-TC per la stadiazione e la valutazione dei recettori della somatostatina.
Trattamento
Come per gli altri NET è multimodale, includendo in primis la chirurgia, con intento radicale quando è possibile o come debulking nelle forme metastatiche, e altre terapie come le loco-regionali (embolizzazione/chemio-emobolizzazione, radiofrequenza, radio-embolizzazione con microsfere) per le metastasi epatiche e la terapia radio-recettoriale con analoghi marcati della somatostatina nelle forme diffuse (9).
Un ruolo fondamentale nel controllo della sindrome è rappresentato dall’utilizzo degli analoghi della somatostatina, efficaci in > 80% dei casi.
Un’attenzione particolare deve essere rivolta alla crisi da VIPoma, che può mettere in pericolo la vita del paziente: deve essere trattata in reparto di terapia intensiva con analoghi della somatostatina anche in infusione continua, corticosteroidi e infusione di liquidi ed elettroliti.
Prognosi
Dipende dalle dimensioni tumorali e dalla presenza ed estensione delle metastasi, con una sopravvivenza media a 5 anni del 60% nei pazienti con metastasi vs 94% nei pazienti senza metastasi.
Bibliografia
- Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev 2004, 25: 458-511.
- Soga J, Yakuwa Y. Vipoma/diarrheogenic syndrome: a statistical evaluation of 241 reported cases. J Exp Clin Cancer Res 1998, 17: 389-400.
- Vagefi PA, Razo O, Deshpande V, et al. Evolving patterns in the detection and outcomes of pancreatic neuroendocrine neoplasms. The Massachusetts General Hospital Experience from 1977 to 2005. Arch Surg 2007, 142: 347-54.
- Peng SY, Li JT, Liu YB, et al. Diagnosis and treatment of VIPoma in China: (case report and 31 cases review) diagnosis and treatment of VIPoma. Pancreas 2004, 28: 93-98.
- Ghaferi-Karen AA, Choinacki A, et al. Pancreatic VIPomas: subject review and one Institutional experience. J Gastrointest Surg 2008, 12: 382-93.
- Smith SL, Branton SA, Avino AJ, et al. Vasoactive intestinal polypeptide secreting islet cell tumors: a 15-year experience and review of the literature. Surgery 1998, 124: 1050-5.
- Nikou GC, Toubanakis C, Nikolaou P, et al. VIPomas: an update in diagnosis and management in a series of 11 patients. Hepatogastroenterology 2005, 52: 1259-65.
- O'Toole D, Grossman A, Gross D, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: biochemical markers. Neuroendocrinology 2009, 90: 194-202.
- Jensen RT, Cadiot G, Brandi ML, et al. ENETS Consensus Guidelines for the Management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 2012, 95: 98-119.
GEP NEN non secernenti
Marco Toaiari1, Elisa Cosaro2, Giuseppe Francia1
1Ospedale Pederzoli, Peschiera del Garda
2MMG ULSS 9 Scaligera
(aggiornato al 1 giugno 2020)
NET NON FUNZIONANTI DEL PANCREAS
Definizione
Le neoplasie neuroendocrine pancreatiche non funzionanti (NF-pNEN) sono un gruppo a comportamento eterogeneo. Il termine “non funzionante” sta ad indicare l'assenza di sintomatologia endocrina correlata. In realtà queste neoplasie sono in grado di sintetizzare molecole ormonali, che però sono inattive o secrete in quantità insufficiente. Prodotti di secrezione tipici, importanti come marcatori diagnostici e prognostici ma privi di effetti biologici, sono la cromogranina A (CgA, 70-100%) e il polipeptide pancreatico (PP) (50-100%).
Epidemiologia
Le NF-pNEN rappresentano la maggioranza delle pNEN (60-70% vs 30-40% delle forme funzionanti) e circa il 12% dei GEP-NET. L'aumento dell'incidenza a cui si è assistito negli ultimi anni è dovuto al riscontro casuale sempre più frequente di piccole neoformazioni pancreatiche in corso di indagini radiologiche eseguite per altre finalità diagnostiche. In accordo con queste osservazioni, studi autoptici hanno riportato una frequenza di NF-pNEN pancreatiche < 1 cm oscillante tra 0.8-10% (1).
La maggior parte delle NF-pNEN sono sporadiche e singole. Le forme familiari e multiple si manifestano nell'ambito della MEN-1, dove si associano con variabile frequenza ad altre malattie endocrine, quali iperparatiroidismo primitivo e adenoma ipofisario. In questo gruppo di pazienti le NF-pNEN costituiscono la neoplasia pancreatica prevalente (dal 19% al 53% dei casi) e la causa più frequente di mortalità. Altra sindrome ereditaria dove si può osservare la presenza di NF-pNEN (13-17% dei casi) è la sindrome di Von Hippel Lindau, caratterizzata da altre neoplasie endocrine (feocromocitoma) e non endocrine (emangioblastoma cerebellare, carcinoma renale). Le pNEN possono essere anche presenti in altre due rare patologie genetiche, la neurofibromatosi di tipo 1 (NF1) e la sclerosi tuberosa (TSC) (2). Al momento non sono conosciuti fattori di rischio ambientali per tali neoplasie. La stragrande maggioranza delle NF-pNEN sono bene/moderatamente differenziate (gradi G1/G2), mentre sono rari i NET G3 o i NEC.
Clinica
Nella maggior parte dei casi le lesioni scoperte incidentalmente con diametro ≤ 2 cm sono benigne o a rischio intermedio e solo il 6% manifesta un comportamento maligno (3). Anche le NF-pNEN associate a MEN 1 con diametro ≤ 2 cm hanno in genere un comportamento poco aggressivo e solo nel 16% dei casi è stato necessario un intervento chirurgico nel corso del follow-up durato 10 anni (4).
Le NF-pNEN diventano clinicamente apparenti quando raggiungono dimensioni tali da interessare gli organi adiacenti (> 5 cm nel 70% delle forme avanzate) o quando hanno causato metastasi, in genere epatiche e/o ossee. La sintomatologia è aspecifica, caratterizzata da dolore addominale (35-78% dei casi), perdita di peso (20-35%), anoressia e nausea (45%); meno frequenti sono l’ittero (17-50%) o una massa palpabile (7-40%). Al momento della diagnosi la prevalenza di metastasi epatiche oscilla tra 32 e 73% (5).
Diagnosi di laboratorio
Attualmente disponiamo di due marcatori nella diagnosi di questi tumori: la cromogranina A (CgA) e l’enolasi neurone-specifica (NSE).
La cromogranina A è una glicoproteina presente nei granuli secretori di tutti i tipi di cellule neuroendocrine e secreta all’esterno durante il comune processo di esocitosi. È elevata nella maggior parte delle pNEN (circa 70%), ma presenta ridotta specificità, potendo essere elevata anche in altri tipi di neoplasie (es. feocromocitoma o carcinoma midollare della tiroide) e in alcune situazioni non oncologiche (ipertensione arteriosa, gastrite atrofica, utilizzo di PPI, insufficienza renale cronica, epatica e cardiaca) (6). Tale molecola correla con il carico di malattia ed è particolarmente sensibile nei pazienti metastatici (7), potendo inoltre essere usata come marcatore di recidiva precoce durante il follow-up. Sebbene recentemente l’utilità clinica della CgA sia stata messa in discussione a causa della bassa specificità e della variabile sensibilità (8), le linee guida ne contemplano ancora l’uso (9).
L’NSE è un enzima glicolitico neurone-specifico, la cui determinazione si è dimostrata particolarmente utile nelle forme tumorali meno differenziate. La sua utilità clinica è comunque relativa, essendo confinata ai rari casi di tumore particolarmente aggressivo (NEC G3).
Diagnosi strumentale
TC e RM rivestono un ruolo prioritario nella diagnostica radiologica.
La TC con mezzo di contrasto resta ancora la metodica di riferimento grazie a fruibilità e diffusione. È in grado di identificare con grande accuratezza sia le lesioni primitive (usualmente ben delimitate, ipervascolarizzate e con vivace enhancement post contrastografico) che quelle secondarie, fornendo inoltre il supporto a procedure bioptiche mirate. Tale metodica, per la riproducibilità, rimane inoltre di prima scelta per il follow-up del paziente e per la valutazione dell’eventuale progressione di malattia.
La RM manifesta sensibilità e specificità sovrapponibili alla TC, con una serie aggiuntiva di vantaggi: non esposizione a radiazioni ionizzanti dei pazienti giovani che necessitano di follow-up in tempi ravvicinati; identificazione di neoplasie pancreatiche ipovascolarizzate (grazie alla possibilità di effettuare scansioni particolari, quali le sequenze DWI); identificazione di metastasi epatiche di piccole dimensioni; definizione più accurata di eventuali lesioni ossee.
L’eco-endoscopia (EUS) è diventata una metodica imprescindibile per lo studio dei pazienti con pNEN. Le lesioni si presentano usualmente tondeggianti, con margini netti, ipoecogene e con presa di contrasto vivace (10). EUS consente inoltre una stadiazione locale accurata (valutazione di linfoadenopatie o infiltrazioni di vasi e organi vicini, distanza dal dotto pancreatico) e la possibilità di eseguire prelievi citologici con ago sottile (FNA) di elevata resa diagnostica. In un recente lavoro la concordanza di Ki-67 tra prelievo ecoendoscopico e pezzo operatorio è risultata dell’84% (11).
Accanto all’imaging “morfologico”, in questi tipi di neoplasia sta assumendo sempre più importanza l’imaging “funzionale”.
La 68Ga-PET/TC è ormai diventata la metodica di riferimento, almeno per quanto riguarda NET G1 e G2, con sensibilità che varia tra 86 e 100% e specificità tra 85 e 100% (12). Tale indagine non solo permette di valutare l’espressione del recettore per la somatostatina tipo 2 (SSTR-2), in previsione di un’eventuale terapia radiometabolica, ma anche di acquisire informazioni prognostiche: l’intensa positività si associa in genere a forme meno aggressive. Da studi di confronto la PET si è anche dimostrata in grado di stimare con più accuratezza il carico di malattia rispetto a TC ed RM, consentendo una migliore diagnosi/stadiazione di malattia (13).
L’altra metodica di imaging funzionale importante, complementare alla Ga-PET è, la 18F-FDG PET/TC, che manifesta scarsa sensibilità per i NET G1 e G2 (attorno al 58%), mentre risulta molto più utile nelle forme meno differenziate (NET G3 e NEC), associandosi a fenotipi più aggressivi e prognosi peggiore. Le più recenti linee guida internazionali raccomandano l’esecuzione di 18F-FDG PET/TC nei NET G3 e NEC per la stadiazione e la stratificazione prognostica, mentre la consigliano nei NET G1 e G2 solo in precisi contesti clinici (es. progressione precoce di malattia, sospetta modifica del Ki67, inefficacia terapeutica) e comunque in un contesto multi-disciplinare (14).
Terapia chirurgica
La terapia di queste neoplasie è in prima istanza chirurgica (almeno per le forme bene o moderatamente differenziate). La via laparoscopica è, se possibile, da preferire, in quanto sembra gravata da minori complicanze.
Ai fini di una corretta strategia terapeutica, oltre che la presenza e l'entità delle metastasi, è di fondamentale importanza la situazione clinica del paziente, la resecabilità o meno della neoplasia, la valutazione anche ripetuta nel tempo dell’attività proliferativa (Ki-67) e l’espressione o meno dei recettori per la somatostatina.
Per quanto riguarda le lesioni ≤ 2 cm, pur non essendoci in merito unanimità di consenso, viene ritenuta in genere giustificata una condotta conservativa, basata su uno stretto follow-up strumentale (ogni tre mesi nel primo anno, e quindi ogni 6 mesi) (15).
Per lesioni pancreatiche > 2 cm localizzate e circoscritte l’intervento chirurgico può essere curativo e deve essere pianificato prendendo in considerazione l’età del paziente, le sue comorbilità e la sede del tumore primitivo.
L’intervento chirurgico sul primitivo è indicato anche nelle neoplasie > 2 cm localmente avanzate ma non metastatiche. In considerazione infatti dell’andamento usualmente indolente di tali neoplasie, sempre più esperienze riportano buoni risultati sulla sopravvivenza globale, soprattutto se l’intervento risulta radicale (R0/R1). L’approccio chirurgico deve comunque essere contestualizzato all’interno di un programma che preveda uno stretto follow-up e un trattamento multimodale (16). Controindicazioni assolute all’intervento sono l’infiltrazione della vena porta complicata dallo sviluppo di cavernoma portale e l’infiltrazione circonferenziale dell’arteria mesenterica superiore.
Nei pazienti con tumori primitivi resecabili ma con metastasi non resecabili le ultime esperienze sembrano suggerire che l’asportazione chirurgica della lesione primitiva può conferire un vantaggio in termini di sopravvivenza (17), anche perché può aumentare l’efficacia di eventuali successivi trattamenti (18).
La chirurgia delle metastasi epatiche resecabili è possibile solo nel 10% dei casi, data la frequente multi-focalità. Può essere presa in considerazione se sono soddisfatti i seguenti criteri (5):
- Ki 67 < 20% su campione bioptico;
- assenza di malattia extra-addominale;
- positività per recettori della somatostatina ad una metodica di imaging funzionale.
Infine, nelle metastasi epatiche diffuse, purchè di dimensioni contenute e con carico tumorale limitato, possono trovare indicazione le terapie loco-regionali quali la (chemio)embolizzazione, l'ablazione con radiofrequenza e la radio-embolizzazione, di cui però mancano tuttora studi prospettici e randomizzati. L'associazione di queste terapie con la PPRT sembra ottenere risultati sinergici, anche se le modalità di integrazione e la successione temporale di queste procedure restano ancora da definire.
Terapia chirurgica nella MEN-1
Nei pazienti affetti da MEN-1, dove le lesioni sono spesso multiple e distribuite in tutto il parenchima pancreatico, l’approccio chirurgico è ancora controverso. In alcuni casi selezionati può essere indicata una chirurgia profilattica, mentre in linea generale la chirurgia dovrebbe essere riservata a NF-pNEN di diametro ≥ 2 cm o con un aumento di dimensioni > 0.5 cm/anno, utilizzando se possibile la tecnica chirurgica che garantisca minori complicanze a lungo termine (resezioni atipiche), data la precoce insorgenza di neoplasie pancreatiche in questi soggetti e la frequente necessità di eseguire interventi multipli (4).
Terapia medica
Per quanto riguarda la terapia farmacologica nelle NF-pNEN non resecabili, gli analoghi della somatostatina (SSA) a lunga durata d'azione (octreotide e lanreotide) rappresentano l'opzione iniziale nelle forme ben differenziate (G1/G2). In un recente studio randomizzato, in doppio cieco e controllato (CLARINET), condotto su 204 pazienti con tumori neuroendocrini entero-pancreatici non funzionanti e metastatici, di grado 1-2 (Ki-67 < 10%), di cui 91 di origine pancreatica, la terapia con lanreotide autogel si è associata a un significativo prolungamento della sopravvivenza libera da progressione di malattia (PFS), indipendentemente dal carico metastatico epatico (19). Sebbene siano utilizzate frequentemente nella pratica clinica dosi elevate di SSA, una recente metanalisi ha mostrato che tale terapia si associa a miglioramento della sindrome clinica ormonale ma con effetti non chiari e univoci sulla sopravvivenza. Nel contesto clinico delle NF-pNEN tale approccio terapeutico non sembra pertanto trovare ancora raccomandazione forte (20). Gli effetti collaterali di tali farmaci sono modesti: flatulenza, dolori addominali, diarrea e nausea, peggioramento del compenso glucidico, colelitiasi e raramente bradicardia (21).
Nei pazienti con NET bene o moderatamente differenziati (G1/G2) l'armamentario farmacologico si è arricchito negli ultimi anni di due nuovi farmaci a bersaglio molecolare: l'everolimus, inibitore di mTOR, e il sunitinib, inibitore tirosin-chinasico. Tali farmaci hanno dimostrato di prolungare in maniera significativa e sovrapponibile la PFS rispetto al placebo, mentre al momento non vi sono dati a favore di un aumento significativo sulla sopravvivenza globale (OS).
L’approvazione di everolimus (EVE) nel trattamento delle pNEN si basa sui risultati dello studio RADIANT-3 (22), dove tale molecola ha mostrato raddoppio della PFS (11 vs 6 mesi) nei tumori G1 e G2, avanzati e in progressione. Studi successivi hanno inoltre evidenziato come tale beneficio si mantenga indipendentemente dalla terapia eseguita in precedenza (23). Esperienze molto recenti (24) suggeriscono un impiego più allargato di EVE anche nelle forme più aggressive (NET G3 con Ki 67 < 55%), ma questi dati incoraggianti devono ancora essere confermati. I principali effetti tossici sono stomatite, diarrea, alterazioni della crasi ematica e iperglicemia. Studi di fase II suggeriscono che l’associazione EVE + SSA possa migliorare la risposta terapeutica (25); mancano tuttavia studi prospettici su tale associazione.
Il sunitinib (SUN), è stato invece approvato grazie ad uno studio del 2011, nel quale il farmaco ha mostrato un raddoppio della PFS (11.4 mesi vs 5.5) nei pazienti con pNEN ben differenziata in progressione. Gli effetti collaterali più significativi sono stati neutropenia (12%), ipertensione (11%) eritrodisestesia palmo-plantare (6%) e disturbi gastrointestinali (6%) (26).
Terapia radiorecettoriale (PRRT)
Negli ultimi anni sta assumendo sempre di più un ruolo importante nella gestione di queste neoplasie o almeno in quelle con elevata espressione SSTR-2, valutata preliminarmente con imaging funzionale (111-In-pentetreotide o ancora meglio 68-Ga-DOTA-peptide PET/TC). Sebbene l’unico studio di fase III prospettico riguardi le NEN del piccolo intestino (NETTER-1, vedi oltre), in alcuni studi retrospettivi sono segnalate significative risposte positive anche nel sottogruppo di pazienti con pNEN, con stabilizzazione di malattia in un’alta percentuale di casi (27,28).
Chemioterapia
La chemioterapia tradizionale nelle forme G1/G2 in progressione si basava sull'associazione 5-FU + streptozotocina/doxorubicina, la cui percentuale di risposta oscilla tra il 35-40% (29). La streptozotocina tuttavia è difficilmente reperibile in Italia e quindi usata raramente, anche perchè mal tollerata.
Negli ultimi anni si sono accumulate una serie di evidenze che hanno mostrato come un regime chemioterapico a base di temozolomide e capecitabina possa ottenere il controllo di malattia in circa l’80% dei casi (30,31). Per pazienti in progressione e con performance status scaduto recentemente è stata proposta la somministrazione di temozolomide metronomica, con buoni risultati e scarsi effetti collaterali (32).
Nelle forme neoplastiche più aggressive (NET G3 o NEC) recenti studi clinici hanno dimostrato la possibilità di una maggiore “sartorializzazione” della chemioterapia. Uno studio scandinavo, infatti, ha mostrato un’importante differenza di risposta alla chemioterapia utilizzando un cut-off del 55% per il Ki67 (33).
Seppur con tutti i limiti degli studi clinici a nostra disposizione, per le neoplasie con Ki 67 > 55% la combinazione cisplatino/etoposide è ancora quella attualmente raccomandata (eventualmente modificata con carboplatino e irinotecano), mentre per le forme con Ki 67 < 55% una chemioterapia efficace sembra l’associazione temozolomide-capecitabina (più simile quindi a quella consigliata per le neoplasie ben differenziate).
Trapianto di fegato
Negli ultimi anni si è fatta strada l’ipotesi trapiantologica come ultimo approccio. Già in passato dati emersi da studi retrospettivi su casistiche limitate suggerivano un beneficio di sopravvivenza in pazienti ben selezionati. Negli ultimi anni nuovi studi hanno fornito ulteriori elementi a favore di questo approccio, che deve essere effettuato in centri altamente specializzati. Al momento il trapianto di fegato potrebbe essere preso in considerazione in pazienti con età < 60 anni, NET G1 o G2, stabile radiologicamente da almeno 6 mesi, assenza di malattia extra-epatica, con primitivo asportato e con interessamento epatico < 50% (34).
Follow-up
Le modalità sono ancora controverse. Le ultime linee guida ENETS forniscono raccomandazioni abbastanza stringenti sulla tempistica dell’imaging morfologica ma più deboli sulla tempistica e l’utilità dell’imaging funzionale. Al momento pertanto si consiglia:
- pazienti con pNET G1 e G2 TC/RM ogni 6-9 mesi (9);
- neoplasie a maggiore aggressività (NEC G3) TC/RM ogni 3 mesi (35).
Per quanto riguarda l’imaging funzionale:
- NET G1 o G2: ricerca dei recettori della somatostatina consigliata ogni 2 anni (o prima se si sospetta una progressione di malattia);
- NETG3/NEC: FDG consigliata solo se l’imaging convenzionale risulta equivoca.
Lavori recenti tuttavia propongono un utilizzo più frequente di tali metodiche funzionali (ogni anno), dato che in ampie casistiche retrospettive tale approccio ha dimostrato di modificare il management del paziente in circa il 75% dei casi (36).
Bibliografia
- Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci 1991, 36: 933-42.
- Capelli P, Martignoni G, et al. Endocrine neoplasm of the pancreas: pathologic and genetic features. Arch Pathol Lab Med 2009, 133: 350-64.
- Bettini R, Partelli S, Boninsegna L, et al. Tumor size correlates with malignancy in non-functioning endocrine tumor. Surgery 2011, 150: 75-82.
- Triponez F, Goudet P, et al. Long term follow-up of Men 1 patients with non-operated small <2 cm non-functional neuroendocrine tumor of the pancreas (NF-PET). 10th Annual Conference for the Diagnosis and Treatment of Neuroendocrine Tumor Disease, 6-8 March 2013: abstr 762.
- Falconi M, Bartsch DK, Eriksson B, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms of digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology 2012, 95: 120-34.
- Modlin IM, Gustafsson BI, et al. Chromogranin A - Biological function and clinical utility in neuroendocrine tumor disease. Ann Surg Oncol 2010, 17: 2427-43.
- Peracchi M, Conte D, Gebbia C, et al. Plasma chromogranin A in patients with sporadic gastroenteropancreatic neuroendocrine tumors or multiple endocrine neoplasia type 1. Eur J Endocrinol 2003, 148: 39-43.
- Marotta V, Zatelli MC, Sciammarella C, et al. Chromogranin A as circulating marker for diagnosis and management of neuroendocrine neoplasm: more flaws than fame. Endocr Relat Cancer 2018, 25: R11-29.
- Falconi M, Eriksson B, et al. ENETS consensus guidelines update for the management of patients with functional pancreatic neuroendocrine tumors and non-functional pancreatic neuroendocrine tumors. Neuroendocrinology 2016, 103: 153-71.
- Kitano M, Kudo M, Yamao K, et al. Characterization of small solid tumors in the pancreas: the value of contrast-enhanced harmonic endoscopic ultrasonography. Am J Gastroenterol 2012, 107: 303-10.
- Di Leo M, Poliani L, Rahal D, et al. Pancreatic neuroendocrine tumors: the role of endoscopic ultrasound biopsy in diagnosis and grading based on the WHO 2017 classification. Dig Dis 2019, 37: 325-33.
- Treglia G, Castaldi P, et al. Diagnostic performance of Gallium-68 somatostatin receptor PET and PET/TC in patients with thoracic and gastroenteropancreatic neuroendocrine tumors: a metanalysis. Endocrine 2012, 42: 80-7.
- Hope TA, Bergsland EK, et al. Appropriate use criteria for somatostatin receptor PET imaging in neuroendocrine tumors. J Nucl Med 2018, 59: 66-74.
- Sundin A, Arnold R, Baudin E, et al. Antibes Consensus Conference partecipants. ENETS consensus guidelines for the standards of care in neuroendocrine tumor: radiological, nuclear and hybrid imaging. Neuroendocrinology 2017, 105: 212-44.
- Gaujoux S, Partelli S, Maire F, et al. Observational study of natural history of small sporadic nonfunctioning pancreatic neuroendocrine tumors. J Clin Endocrinol Metab 2013, 98: 4784–9.
- Kleine M, Schrem H, et al. Extended surgery for advanced pancreatic endocrine tumors. Br J Surg 2012, 99: 88-94.
- Partelli S, Cirocchi L, et al. A systematic review and metanalysis on the role of palliative primary resection for pancreatic neuroendocrine neoplasm with liver metastasis. HPB 2018, 20: 197-213.
- Bertani E, Fazio N, et al. Resection of the primary tumor followed by peptide receptor radionucleotide therapy as upfront strategy for treatment of G1-G2 pancreatic neuroendocrine tumors with unresecable liver metastases. Ann Surg Oncol 2016, 23 suppl 5: S981-9.
- Caplin ME, Pavel M, Cwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014, 371: 224-33.
- Chan DL, Ferone D, Albertelli M, et al. Escalated-dose somatostatin analogues for antiproliferative effect in GEP-NETS: a systematic review. Endocrine 2017, 57: 366-75.
- Oberg K, Kvols S, Caplin M, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol 2004, 15: 966-73.
- Yao JC, Shah MH, Ito T, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 514-23.
- Buzzoni R, Carnaghi C, et al. Impact of prior therapies on everolimus activity: an exploratory analysis of RADIANT-4. Onco Targets Ther 2017, 10: 5013-30.
- Panzuto F, et al. Everolimus in pancreatic neuroendocrine carcinomas G3. Pancreas 2017, 46: 302-5.
- Capdevilla J, Sevilla I, et al. Evaluation of the efficacy and safety of lanreotide in combination with targeted therapies in patients with neuroendocrine tumors in clinical practice: A retrospective cross-sectional analysis. BMC Cancer 2015, 15: 495.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 501-13.
- Kim SJ, Pak K, Koo PJ, et al. The efficacy of 177Lu-labelled peptide receptor radionucleotide therapy in patients with neuroendocrine tumors: a metanalysis. Eur J Nucl Med Mol Imaging 2015, 42: 1964-70.
- Brabander T, van der Zwan WA, et al. Long term efficacy, survival, and safety of [177Lu-DOTA0-Tyr3] octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin Cancer Res 2017, 23: 4617-24.
- Kouvaraki MA, Ajani JA, Hoff P, et al. Fluorouracil, doxorubicin and streptozocin in the treatment of patients with locally advanced pancreatic endocrine carcinoma. J Clin Oncol 2004, 22: 4762-71.
- Strosberg JR, Fine RL, Choi J, et al. First line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinoma. Cancer 2011, 117: 268-75.
- Spada F, Antonuzzo L, Marconcini R, et al. Chemotherapy with capecitabine and temozolomide (CAP-TEM) in patients with advanced neuroendocrine neoplasm: an Italian multicenter retrospective analysis. J Clin Oncol 2015, 33 Suppl: abstr e15174.
- Tafuto S, Von Arx C, et al. Safety and activity of metronomic temozolomide in second-line treatment of advanced neuroendocrine neoplasm. J Clin Med 2019, 15: 1224.
- Sorbye H, Welin S, Langer SW, et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal poorly differentiated neuroendocrine carcinoma: the NORDIC NEC study. Ann Oncol 2013, 24: 152-60.
- Mazzaferro V, Sposito C, et al. The long term benefit of liver transplantation for hepatic metastasis from neuroendocrine tumors. Amer J Transplant 2016, 16: 2892-902.
- Garcia-Carbonero R, Sorbye B, et al. ENETS consensus guidelines for high-grade gastroenteropancreatic neuroendocrine tumors and neuroendocrine carcinomas. Neuroendocrinology 2016, 103: 186-94.
- Merola E, Pavel ME, et al. Functional imaging in the follow-up of enteropancreatic neuroendocrine tumors: clinical usefulness and indication. J Clin Endocrinol Metab 2017, 102: 1486-94.
NET GASTRICI NON FUNZIONANTI
Classificazione, epidemiologia e patologia
I NET dello stomaco (carcinoidi gastrici) originano dalle cellule enterochromaffin-like (ECL) della mucosa oxintica del fondo e corpo gastrico. Rappresentano il 23% di tutte le neoplasie del tratto gastro-intestinale (1).
Se ne distinguono tre tipi.
Tipo 1 (70-80%), associato a gastrite cronica atrofica del corpo e fondo, può riconoscere eziologia autoimmune o in rari casi sorgere su una infezione cronica da Helicobacter Pylori (2). L’ipergastrinemia, secondaria all’acloridria, è il movente eziopatogenetico di questa forma, in quanto fattore di stimolo alla proliferazione delle ECL. Le lesioni sono in genere piccole (< 1 cm) e multiple, di aspetto spesso polipoide, confinate alla mucosa e sottomucosa. L’indice proliferativo è in genere < 2% (NET G1). Sono molto rare (< 2.5% dei casi) le metastasi a linfonodi regionali e fegato. Marcatore immunitario della gastrite atrofica è la positività degli anticorpi anti-cellule parietali gastriche (APCA). È possibile la coesistenza di altre malattie autoimmuni, quali tiroidite autoimmune e diabete mellito tipo 1, che configurano il quadro della sindrome poliendocrina tipo III. Tipica è l'associazione con anemia perniciosa, da malassorbimento di vitamina B12 per deficit del fattore intrinseco.
Tipo 2 (5-8%) è anch’esso gastrina-dipendente. L’ormone è secreto in maniera autonoma da NET duodenali e/o pancreatici (sindrome di Zollinger Ellison). La mutazione del gene della MEN-1 sembra contribuire all'insorgenza di queste neoplasie. Le lesioni sono multiple, di dimensioni < 1 cm e limitate a mucosa-sottomucosa (Grado 1/2, Ki-67 < 2%). Esse tuttavia manifestano un comportamento più aggressivo del tipo 1, con una frequenza di metastasi linfonodali fino al 30% ed epatiche fino al 10%.
Tipo 3 (15-20%), non è associato a ipergastrinemia ed è singolo, sporadico, di dimensioni > 1 cm nel 70% dei casi. Il comportamento è aggressivo (usualmente NET G3): la neoplasia invade la parete nel 75% dei casi e metastatizza ai linfonodi e al fegato fino al 70% dei casi.
È stata infine descritta una forma scarsamente differenziata molto rara (nota come carcinoide tipo 4), localizzata in sede antrale, di grandi dimensioni (> 4 cm), caratterizzata da comportamento francamente maligno (Grado 3, Ki-67 > 15%) e prognosi infausta (3).
Quadro clinico
La sintomatologia del tipo 1 è aspecifica. Nella maggior parte dei casi la gastrite atrofica autoimmune è asintomatica o associata a vaghi disturbi dispeptici e viene diagnosticata per sintomi correlati al malassorbimento di ferro (anemia sideropenica) e vitamina B12. Possibile, anche se rara, è la diarrea da colonizzazione batterica del piccolo intestino conseguente all’acloridria.
Nel tipo 2 possono essere prevalenti i sintomi correlati all'ipergastrinemia primitiva, quali i dolori addominali simil-ulcerosi e la diarrea.
Infine, nei tipi 3 e 4 la sintomatologia può essere assai grave, caratterizzata da dolore addominale, diarrea, emorragia gastrica, perdita di peso ed episodi di ostruzione gastrica con vomito.
La sindrome da carcinoide, di carattere atipico e di tipo istaminergico (flushing, edema facciale, lacrimazione, rinorrea) è rarissima (< 1%) e si associa in genere al tipo 3.
Diagnosi
La diagnosi avviene spesso accidentalmente in corso di esami endoscopici eseguiti per altri motivi.
L’EGDS permette di definire l’eventuale multi-focalità delle lesioni, di eseguire biopsie per la conferma istologica e per valutare la presenza di ulteriori lesioni mucosali.
Nelle lesioni > 1 cm è indicata l’eco-endoscopia (EUS), che permette di chiarire il grado di infiltrazione di parete e la presenza di eventuali lesioni linfonodali.
In casi selezionati e nelle forme più aggressive può essere utile l’imaging morfologico (TC o RM) o funzionale (PET con Ga-68).
Trattamento
È controverso e dipende dal tipo di NET e dalle dimensioni e numero dei polipi.
Le lesioni < 1 cm possono essere controllate periodicamente (EGDS ogni 12 o 24 mesi) o sottoposte a rimozione endoscopica in casi selezionati.
Nelle lesioni di tipo 1 o tipo 2 > 1 cm è consigliabile eseguire EUS prima della dissezione sottomucosa (ESD) o della resezione mucosale endoscopica (EMR). Non esistono al momento studi di confronto tra le due metodiche, che pertanto possono essere utilizzate indifferentemente tenendo conto dell’esperienza dell’operatore.
L'analogo della somatostatina si è dimostrato in grado di far regredire i polipi in serie limitate di pazienti, ma vista l’assenza di studi prospettici rispetto alla sorveglianza endoscopica il suo impiego non è ad oggi raccomandato di routine per i NET G1. Il suo utilizzo in questo tipo di neoplasie può essere preso in considerazione in casi selezionati che presentano frequenti recidive, nelle forme multiple (> 6) e dopo numerose bonifiche endoscopiche (4).
Nei tumori di tipo 2 è sufficiente in genere l'escissione locale (ESD o EMR), mentre l'analogo della somatostatina può essere indicato per il controllo della sindrome di Zollinger-Ellison (insieme ovviamente ai PPI) e nei polipi multipli.
Le lesioni di tipo 3, che manifestano un comportamento biologico simile a quello dell'adenocarcinoma gastrico, richiedono invece un trattamento chirurgico radicale (gastrectomia + dissezione linfonodale) ed eventualmente chemioterapia neoadiuvante con schemi a base di platino (5).
Bibliografia
- Niederle MB, Hackl M, Kaserer K, et al. Gastroenteropancreatic neuroendocrine tumours: the current incidence and staging based on the WHO and European Neuroendocrine Tumour Society classification: an analysis based on prospectively collected parameters. Endocr Relat Cancer 2010, 17: 909–18.
- Yamaguchi E, Iwasa T, et al. Gastric neuroendocrine tumor with hypergastrinemia following type B chronic atrophic gastritis: a case report. Nihon Shokakibyo Gakkai Zasshi 2017, 114: 248-55.
- Kulke MH, Anthony LB, Bushnell DL, et al. NANETS treatment guidelines: well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas 2010, 39: 735-52.
- Delle Fave G, O’Toole D, et al. ENETS consensus guidelines update for gastroduodenal neuroendocrine neoplasm. Neuroendocrinology 2016, 103: 119-24.
- O' Toole D, Delle Fave G, Jensen RT. Gastric and duodenal neuroendocrine tumors. Best Pract Res Clin Gastroenterol 2012, 26: 719-35.
NET NON FUNZIONANTI DEL DIGIUNO-ILEO
CLASSIFICAZIONE, EPIDEMIOLOGIA E PATOLOGIA
Le NEN intestinali derivano dalle cellule neuroendocrine dell’intestino che producono serotonina. Sono le più comuni NEN gastro-intestinali (30%) e rappresentano il 35% delle neoplasie del piccolo intestino. La loro prevalenza oscilla da un minimo di 0.32/100.000 in Inghilterra (1) a un massimo di 1.12/100.000 in Svezia (2), con lieve prevalenza nel sesso maschile e negli afro-americani (3). L'età media alla diagnosi si colloca verso la fine della 5° decade, con prevalenza nella 7° decade (4).
Nella maggior parte dei casi sono localizzate nel tratto terminale del tenue e nel 25% dei casi possono essere multi-focali.
Dal punto di vista del comportamento biologico, queste neoplasie sono nella maggior parte dei casi di basso grado (G1 o G2) e spesso indolenti, mentre i NET G3 sono di riscontro eccezionale.
La prognosi dipende sia dallo stadio (TNM) che dal grading (Ki-67). Considerando lo stadio alla diagnosi, il tasso di sopravvivenza a 5 anni è del 100% per gli stadi I e II, del 97.1% per lo stadio III e dell’84.8% per lo stadio IV (5). Fattori prognostici indipendenti sono la presenza di metastasi linfonodali, il carico tumorale epatico e la presenza di metastasi extra-addominali.
Non sono ancora state definitivamente accertate alterazioni genetiche specifiche nelle NEN del piccolo intestino; tuttavia, recenti studi hanno dimostrato la presenza di alcune associazioni familiari che potrebbero suggerire una predisposizione genetica (6,7).
CLINICA
Le forme non funzionanti di NEN intestinali (70%) non si associano per definizione alla sindrome carcinoide e sono spesso diagnosticate già in fase metastatica (48% dei casi) o localmente avanzata (36% dei casi). Il sintomo più frequente descritto nelle casistiche multicentriche è il dolore addominale diffuso, che può essere sottovalutato per anni ed attribuito erroneamente a colon irritabile (8). Col progredire della malattia il dolore addominale può diventare più intenso e di tipo colico (per complicanze ostruttive o ischemiche), associandosi a nausea e vomito, fino al classico quadro di occlusione intestinale. Altri sintomi aspecifici sono astenia e, raramente, febbre di origine sconosciuta. Occasionalmente tali tumori si possono manifestare con melena o rettorragia per sanguinamento enterico o con un quadro di ittero ingravescente, se presenti lesioni epatiche diffuse.
DIAGNOSI
Gli strumenti diagnostici del laboratorio sono limitati, potendo contare sostanzialmente solo sulla cromogranina A, con i limiti di sensibilità e specificità già descritti.
Imaging morfologico
Le metodiche radiologiche convenzionali (TC e RM) giocano un ruolo fondamentale nella diagnosi e stadiazione di queste neoplasie, essendo provviste di elevata specificità e sensibilità. Tuttavia, il problema diagnostico della ricerca del primitivo in presenza di secondarismi di non chiara origine rimane ancora frequente, in relazione alla capacità metastatizzante di lesioni anche molto piccole (< 1 cm), di difficile identificazione. In tale contesto clinico recenti studi pongono un’attenzione particolare all’entero-TC, che ha dimostrato una sensibilità diagnostica che varia dall’86 al 100% (9,10), soprattutto per lesioni > 1 cm, e che può essere di grande aiuto dove le metodiche più “tradizionali” sono negative o di non univoca interpretazione.
Altre metodiche diagnostiche sono la colonscopia (utile nel caso in cui la neoplasia prolassi attraverso la valvola ileo-cecale) o altre più complesse e meno usufruibili, quali la video-capsula o l’enteroscopia a doppio pallone (11).
Imaging funzionale
Negli ultimi anni ha assunto sempre più importanza nella gestione di questi tipi di neoplasia. La 68Ga-PET/TC ormai è diventata una metodica fondamentale, in quanto raggiunge livelli di sensibilità del 90-95%, sia per quanto riguarda la localizzazione del tumore primitivo, sia per la quantificazione del carico di malattia (secondarismi linfonodali, epatici e ossei). Tale indagine risulta fondamentale sia nella fase diagnostica sia nel follow-up ed è fortemente raccomandata in tutti i casi di NEN G1 o G2, soprattutto in presenza di piccole lesioni digiuno-ileali, così come in fase pre-operatoria, per escludere la presenza di metastasi a distanza non individuate con altre metodiche radiologiche convenzionali (12). La PET con 18F-FDG risulta invece una metodica in generale meno utile, essendo indicata solo nei rari casi di lesioni poco differenziate (NEN G3), dove ha una valenza prognostica oltre che diagnostica.
TERAPIA CHIRURGICA
Sulla base dell'estensione della malattia e del contesto clinico, l'intervento chirurgico può essere curativo o avere un significato solo palliativo. L’approccio laparoscopico non è sempre attuabile date la possibile infiltrazione tumorale e la reazione desmoplastica spesso associata. La scelta della tecnica chirurgica dipende, quindi, dall’esperienza dell’operatore e dall’estensione di malattia.
Chirurgia nel paziente non metastatico
Per le NEN del piccolo intestino resecabili e in assenza di metastasi a distanza la terapia è sempre chirurgica e consiste nella resezione del tratto intestinale coinvolto associata ad ampia linfadenectomia, che spesso coinvolge la radice del mesentere. Tale procedura ha dimostrato in ampie casistiche di migliorare significativamente la sopravvivenza a lungo termine (13,14). Qualora la neoplasia coinvolga l’ileo terminale, è spesso necessario eseguire anche emicolectomia destra, con lo scopo di ottenere la maggiore radicalità chirurgica. È raccomandata la colecistectomia a scopo profilattico in corso di intervento, in special modo nei pazienti candidati a terapia con SSA, per prevenire lo sviluppo di calcolosi (15).
Chirurgia del paziente metastatico
In questa situazione si distinguono essenzialmente 3 situazioni:
- paziente con presenza di lesioni epatiche sincrone e resecabili, in cui l’intervento chirurgico sul duplice sito ha dimostrato beneficio clinico evidente (16);
- paziente con metastasi non resecabili ma sintomatico (occlusione o sanguinamento intestinale), in cui l’intervento chirurgico ha sostanzialmente finalità palliative;
- pazienti asintomatici ma con metastasi non resecabili, in cui l’intervento chirurgico sul tumore primitivo appare discutibile. In questo contesto l’approccio chirurgico sul primitivo è contemplato dalle recenti linee guida (17), sebbene fortemente dibattuto visti i risultati contrastanti nelle diverse casistiche (18,19).
Data la complessità di queste situazioni, l’opzione chirurgica implica un’attenta selezione dei pazienti e dei centri chirurgici cui fare riferimento, che devono essere caratterizzati da un elevato volume di interventi e dalla possibilità di una gestione multi-disciplinare.
In presenza di metastasi epatiche diffuse possono trovare indicazione le terapie loco-regionali, quali (chemio)embolizzazione e ablazione con radiofrequenza, per cui però mancano tuttora studi prospettici randomizzati.
Da ultimo in pazienti selezionati con metastasi epatiche diffuse si può considerare l’approccio trapiantologico, da effettuare in casi specifici e in centri altamente specializzati (vedi paragrafo delle NEN pancreatiche).
TERAPIA FARMACOLOGICA
Gli analoghi della somatostatina (SSA) octreotide e lanreotide rappresentano la terapia medica elettiva dei GEP-NET, sia funzionanti che non funzionanti. Anche se già negli anni precedenti molti studi retrospettivi avevano segnalato il possibile effetto anti-proliferativo di tali farmaci, il loro utilizzo si è definitivamente affermato nella pratica clinica dopo la pubblicazione degli studi PROMID e CLARINET.
Nello studio PROMID (fase III, randomizzato vs placebo in doppio cieco) condotto su 85 pazienti con tumori neuroendocrini del midgut ben differenziati, metastatici e in progressione, l’octreotide LAR (30 mg ogni 4 settimane) ha dimostrato di raddoppiare il tempo alla progressione della malattia (14.3 mesi vs 6 mesi) nelle forme con tumore primitivo resecato e con basso carico metastatico epatico (< 10%) (20).
Nello studio CLARINET (fase III, randomizzato vs placebo, in doppio cieco) condotto su 204 pazienti con GEP-NET di grado 1 e 2 (Ki-67 < 10%), non funzionanti e metastatici, lanreotide autogel (120 mg ogni 4 settimane) ha ottenuto un significativo prolungamento della sopravvivenza libera da progressione (PFS), indipendentemente dal carico metastatico epatico (21).
Farmaci a bersaglio molecolare
Sebbene al momento attuale nessun farmaco a bersaglio molecolare sia registrato in Italia per la terapia delle NEN del piccolo intestino, everolimus ha dato prova di efficacia in 2 importanti studi.
Il RADIANT-2 è uno studio prospettico randomizzato di fase III, in doppio cieco, placebo-controllato, che ha valutato l’efficacia di everolimus 10 mg/die in pazienti con sindrome carcinoide associata a NEN avanzata, in progressione e trattati con octreotide LAR 30 mg ogni 4 settimane. Lo studio ha dimostrato un miglioramento della PFS mediana (16.4 vs 11.3 mesi), che tuttavia non ha raggiunto per poco la significatività statistica. Sebbene lo studio in senso stretto sia da considerarsi negativo, il beneficio clinico era comunque presente (22).
Il RADIANT-4 è uno studio prospettico, randomizzato, di fase III, internazionale, multicentrico, in doppio cieco, volto a valutare efficacia e sicurezza di everolimus 10 mg/die contro placebo in pazienti con NET G1-G2 del tratto gastro-intestinale o polmonare. I NET del piccolo intestino rappresentavano il 32% della popolazione arruolata. La PFS mediana è risultata di 11 mesi nei trattati con everolimus vs 3.9 mesi nel gruppo placebo (23).
In base ai risultati del RADIANT-4, FDA e EMA hanno approvato everolimus nel trattamento delle NEN a partenza gastro-intestinale e polmonare, mentre in Italia il farmaco è prescrivibile nei NET ben differenziati del tratto intestinale o del polmone secondo la Legge 648/96 (lista farmaci malattie rare).
Gli effetti avversi di grado 3 e 4 sono stati infrequenti in questi studi: stomatite (9%), diarrea (7%), infezioni (7%), anemia (4%), fatigue (3%) e iperglicemia (3%).
Chemioterapia
Nei NET G1 e G2 non vi sono attualmente evidenze di chiara efficacia delle terapie citotossiche. Le mono-chemioterapie hanno mostrato tassi di risposta obiettiva < 15% e pertanto vengono utilizzate solo in pazienti pre-trattati o in caso di condizioni cliniche generali scadute. I regimi poli-chemioterapici hanno mostrato, invece, una maggiore attività, anche se ad oggi non esistono schemi terapeutici ben codificati. Al netto delle evidenze scientifiche, schemi contenenti fluoropirimidine (5-FU o capecitabina) e oxaliplatino sembrano dare il maggior tasso di risposta con tossicità accettabile (24), mentre la temozolomide, molto efficace nelle NEN pancreatiche, ha prodotto scarsi risultati sui primitivi intestinali (25).
Per i rarissimi NET G3 (soprattutto Ki67 > 55%) può essere ragionevole uno schema a base di cisplatino ed etoposide (o irinotecano), simile a quello impiegato per le neoplasie a partenza pancreatica.
Dopo una chirurgia con intento curativo non è indicata alcuna terapia adiuvante, così come non vi è alcun ruolo per eventuali terapie mediche neo-adiuvanti.
TERAPIA RADIORECETTORIALE
La terapia radiometabolica è una forma di radioterapia sistemica che permette di trasferire i radionuclidi all’interno delle cellule esprimenti i recettori per la somatostatina. Sono stati pubblicati molti studi retrospettivi, prospettici e casi clinici sull’efficacia di tale approccio (26). Con lo studio NETTER-1 la terapia radiometabolica con Lutezio-177 si è imposta definitivamente con livello di evidenza elevato in questi tipi di neoplasia (27). In questo importante studio di fase III, multicentrico, randomizzato, in aperto, la terapia radiometabolica con Lutezio 177-DOTA-TATE è stata confrontata con SSA ad alte dosi (octreotide LAR 60 mg ogni 4 settimane) su 229 pazienti affetti da NEN del piccolo intestino non resecabili, metastatiche e in progressione con l’analogo freddo a dosi convenzionali. La percentuale stimata di PFS a 20 mesi è stata del 65.2% con la terapia radiorecettoriale e del 10.8% nel gruppo di controllo, con una riduzione del rischio di progressione del 79% nei pazienti sottoposti a terapia con Lutezio, a fronte di effetti collaterali ematologici e renali modesti.
FOLLOW-UP
Nei pazienti con NET G1 o G2 sottoposti ad intervento curativo i controlli dovrebbero essere effettuati ogni 6-12 mesi, ad eccezione dei NEC G3 dove il follow-up deve essere più ravvicinato (ogni 3 mesi). Nei pazienti con persistenza tumorale a bassa velocità di crescita il miglior approccio sembra un follow-up semestrale (17).
BIBLIOGRAFIA
- Ellis L, Shale MJ, Coeman MP. Carcinoid tumors of the gastrointestinal tract: trends in incidence in England since 1971. Am J Gastroenterol 2010, 105: 2563-9.
- Landerholm K, Falkmer S, Jarhult J. Epidemiology of small bowel carcinoids in a defined population. World J Surg 2010, 34: 1500-5.
- Yao JC, Hassan M, Phan, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008, 26: 3063-72.
- Srirajaskanthan R, Ahmeds A, Prachialias A, et al. ENETS TNM staging predicts prognosis in small bowel neuroendocrine. ISRN Oncol 2013, 2013: 420795.
- Jann H, Roll S, Couvelard A, et al. Neuroendocrine tumors of midgut and hindgut origin: tumor-node-metastasis classification determines clinical outcome. Cancer 2011, 117: 3332–41.
- Hassan MM, Phan A, Li D, et al. Family history of cancer and associated risk of developing neuroendocrine tumors: a case-control study. Cancer Epidemiol Biomarkers Prev 2008, 17: 959–65.
- Järhult J, Landerholm K, Falkmer S, et al. First report on metastasizing small bowel carcinoids in first-degree relatives in three generations. Neuroendocrinology 2010, 91: 318–23.
- Niederle MB, Niederle B. Diagnosis and treatment of gastroenteropancreatic neuroendocrine tumors: current data on a prospectively collected, retrospectively analyzed clinical multicenter investigation. Oncologist 2011, 16: 602–13.
- Soyer P, Dohan A, et al. Carcinoid tumors of the small bowel: evaluation with 64-section CT enteroclysis. Eur J Radiol 2013, 82: 943-50.
- Kamaoui I, De Luca V, et al. Value of CT-enteroclysis in suspected small-bowel carcinoid tumors. Am J Roentgenol 2010, 194: 629-33.
- Pape UF, Perren A, Niederle B, et al. ENETs consensus guidelines for the management of patients with neuroendocrine neoplasms from jejuno-ileum and the appendix including goblet cell carcinomas. Neuroendocrinology 2012, 95: 135-56.
- Bozkurt MF, Virgolini I, et al. Guideline for PET/TC imaging of neuroendocrine neoplasms with 68Ga-DOTA-coniugated somatostatin receptor targeting peptides and 18F-DOPA. Eur J Nucl Med Mol Imaging 2017, 44: 1588-601.
- Han SL, Cheng J, Zhou HZ, et al. Surgically treated primary malignant tumor of small bowel: a clinical analysis. World J Gastroenterol 2010, 16: 1527–32.
- Landry CS, Lin HY, Phan A, et al. Resection of at-risk mesenteric lymph nodes is associated with improved survival in patients with small bowel neuroendocrine tumors. World J Surg 2013, 37: 1695–700.
- Eriksson J, Stalberg P, Nilsson A, et al. Surgery and radiofrequency ablation for treatment of liver metastases from midgut and foregut carcinoids and endocrine pancreatic tumors. World J Surg 2008, 32: 930–8.
- Schindl M, Kaczirek K, Passler C, et al. Treatment of small intestinal neuroendocrine tumors: is an extended multimodal approach justified? World J Surg 2002, 26: 976–84.
- Niederle B, Pape UF, et al. ENETS consensus guidelines update for neuroendocrine neoplasms of the jejunum and ileum. Neuroendocrinology 2016, 103: 125-38.
- Capurso G, Rinzivillo M, et al. Systematic review of resection of primary midgut carcinoid tumors in patients with unresecable liver metastases. Br J Surg 2012, 99: 1480-6.
- Daskalakis K, Karakatsanis A. Association of a prophylactic surgical approach to stage IV small intestinal neuroendocrine tumors with survival. JAMA Oncol 2018, 4: 183-9.
- Rinke A, Muller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol 2009, 27: 4656-63.
- Caplin ME, Pavel M, Cwikla JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014, 371: 224-33.
- Pavel ME, Hainsworth JD, et al. RADIANT-2 study group. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumors associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet 2011, 378: 2005-12.
- Yao JC, Fazio N, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumors of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet 2016, 387: 968-77.
- Bajetta E, Catena L, et al. Are capecitabine and oxaliplatin (XELOX) suitable treatment for progressing low-grade and high-grade neuroendocrine tumors? Cancer Chemother Pharmacol 2007, 59: 637-42.
- Kulke MH, Stuart L, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol 2006, 24: 401-6.
- Brabander T, van der Zwan WA, et al. Long term efficacy, survival, and safety of [177Lu-DOTA0-Tyr3] octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin Cancer Res 2017, 23: 4617-24.
- Strosberg J, El-Haddad G, Wolin E, et al. NETTER-1 trial investigators. Phase 3 Trial of 177Lu-Dotatate for Midgut Neuroendocrine Tumors. N Engl J Med 2017, 376: 125-35.
NET NON FUNZIONANTI DELL’APPENDICE
Epidemiologia
Le neoplasie neuroendocrine dell’appendice sono relativamente frequenti, con incidenza oscillante tra 0.15-0.6/100.000/anno (1) e riscontro in circa lo 0.3-0.5% delle appendicectomie effettuate per altre cause.
La prevalenza è maggiore nel sesso femminile (rapporto F:M = 2:1).
La neoplasia si può sviluppare in qualunque epoca della vita, compresa l'età infantile, con mediana intorno alla 4° decade (2).
La prognosi usualmente è eccellente qualora il tumore sia asportato in uno stadio precoce (sopravvivenza 95-100% a 5 anni), mentre nei rari casi di tumore avanzato la sopravvivenza a 5 anni raggiunge il 25% circa (3).
Sono stati identificati due sottotipi di NEN dell'appendice:
- tipico: deriva dalle cellule neuroendocrine subepiteliali situate nella lamina propria;
- a “goblet cells” (cellule caliciformi mucipare): sono tumori assai rari (0.01-0.05/100.000) a fenotipo misto, con parziale differenziazione neuroendocrina e ghiandolare mucinosa (4). Si possono quindi considerare come MiNEN (neoplasie neuroendocrine miste) e negli stadi avanzati (5) il potenziale maligno è maggiore di quello del classico NET appendicolare, con comportamento clinico più simile a quello dell'adenocarcinoma.
Clinica e terapia
Quasi tutte queste neoplasie sono clinicamente silenti, scoperte come reperto occasionale in appendici asportate chirurgicamente per vari motivi. La sindrome carcinoide è estremamente rara e dovrebbe far sorgere il sospetto di una diffusione metastatica. In qualche caso si può avere dolore/senso di peso in fossa iliaca destra.
Le dimensioni sono in genere piccole (< 1 cm) e solo raramente superano il diametro di 2 cm. La maggior parte delle lesioni (70%) è localizzata all'estremità dell'appendice, con un’attività proliferativa in genere bassa. Le metastasi, molto rare, interessano in genere i linfonodi; assai meno frequenti sono le metastasi a distanza.
La diagnosi viene quindi effettuata molto spesso in corso di laparotomia o laparoscopia, anche se talvolta può essere sospettata grazie all'ecografia pre-operatoria. Le indagini radiologiche convenzionali (TC e RM) possono essere utili in caso di riscontro accidentale pre-operatorio per la stadiazione addominale della neoplasia. Le metodiche scintigrafiche (68-Ga-PET) dovrebbero essere riservate ai casi in cui si sospettino localizzazioni a distanza.
La terapia è sostanzialmente chirurgica e dipende essenzialmente da dimensione, invasione e sede precisa della neoplasia (6):
- per neoplasie < 1 cm la resezione appendicolare è usualmente sufficiente a garantire una resezione completa, vista l’eccezionalità del rischio metastatico;
- per neoplasie tra 1 e 2 cm il rischio metastatico arriva fino al 25%, per cui la scelta terapeutica è più dibattuta. In generale, oltre all’appendicectomia, l’intervento dovrebbe prevedere emicolectomia destra qualora fossero presenti linfoangio-invasione, localizzazione alla base appendicolare, infiltrazione del meso-appendice per più di 3 mm ed esame istologico compatibile con NET G2 (7);
- per neoplasie > 2 cm o macroscopicamente invasive, è raccomandata una resezione più ampia, che comprende il colon di destra ed eventualmente gli organi coinvolti, in quanto la frequenza delle metastasi arriva anche al 40%.
Approcci farmacologici e oncologici, raramente necessari visto la prognosi eccellente di queste neoplasie, sono comunque comuni con le linee guida delle NEN intestinali e sono riservati a pazienti con malattia avanzata.
Follow-up
Lesioni < 1 cm asportate radicalmente (con o senza emicolectomia) e lesioni tra 1 e 2 cm sottoposte a chirurgia radicale: non è necessario follow-up.
Neoplasia tra 1 e 2 cm, in cui le caratteristiche cliniche del paziente o del tumore rappresentino fattori di rischio particolari (es. metastasi linfonodali, istologia aggressiva) e lesioni > 2 cm: è raccomandato follow-up con TC o RM ogni 6-12 mesi, sebbene non validato da studi clinici (6).
Bibliografia
- Yao JC, Hassan M, Phan, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008, 26: 3063-72.
- Carr NJ, Sobin LH. Neuroendocrine tumors of the appendix. Semin Diagn Pathol 2004, 93: 108-19.
- Connor SJ, Hanna GB, Frizelle FA. Appendiceal tumors: retrospective clinicopathologic analysis of appendiceal tumors from 7970 appendectomies. Dis Colon Rectum 1998, 41: 75-80.
- Kanthal R, Saxena A, Kanthal SC. Goblet cells carcinoids of the appendix: immunophenotype and ultrastructural study. Arch Pathol Lab Med 2001, 125: 386-90.
- Butler JA, Houshiar A, Lin F, Wilson SE. Goblet cell carcinoid of the appendix. Am J Surg 1994, 168: 685-7.
- Pape UF, Niederle B, et al. ENETS Consensus Guidelines for Neuroendocrine Neoplasms of the Appendix (Excluding Goblet Cell Carcinomas). Neuroendocrinology 2016, 103: 144–52.
- Syracuse DC, Perzin KH, Price JB, et al. Carcinoid tumors of appendix. Mesoappendiceal extension and nodal metastases. Ann Surg 1979, 190: 58-63.
NEN NON FUNZIONANTI DEL COLON-RETTO
Le NEN del colon hanno un’incidenza di circa 0.2 casi/100.000/anno e rappresentano circa il 17% delle NEN gastro-intestinali (1). Sembrano più frequenti nell’etnia afro-americana e hanno età media alla diagnosi di circa 60 anni.
Vista l’asintomaticità di tali neoplasie nelle fasi precoci, si riscontrano secondarismi nel 30% dei casi, con una sopravvivenza generale a 5 anni del 50% (2).
Il retto rappresenta la seconda sede più frequente di NEN gastro-intestinale (27.4%) dopo il piccolo intestino, con incidenza di 0.86 casi/100.000/anno. L’età media alla diagnosi è 56 anni e questa localizzazione sembrerebbe più frequente nella popolazione asiatica (3). La prognosi è in genere favorevole, con una sopravvivenza a 5 anni dell'88% circa nelle forme localizzate (4), che rappresentano la maggioranza (78%). La presenza di metastasi ai linfonodi regionali o a distanza riduce la percentuale di sopravvivenza a 5 anni, rispettivamente al 41% e al 25%. La sindrome carcinoide è estremamente rara.
Nella maggioranza dei casi le NEN del colon-retto sono asintomatiche (5) e non infrequentemente il riscontro è occasionale, nel corso di un esame endoscopico eseguito per altri motivi o per screening. Quando presenti, i sintomi più frequenti sono tenesmo, rettorragia, diarrea e perdita di peso, mentre è rara l'occlusione intestinale dato il calibro del retto e del colon. Frequentemente tali tumori vengono diagnosticati nel corso di accertamenti per il riscontro di secondarismi epatici da primitivo sconosciuto.
La tecnica diagnostica d’elezione è la colonscopia. Tuttavia, recenti studi hanno mostrato che in pazienti anziani e fragili o in presenza di colonscopie di difficile esecuzione e in caso di pulizia incompleta, la colonscopia virtuale tramite TC multistrato rappresenta una tecnica molto utile, non invasiva, con sensibilità sovrapponibile alla colonscopia tradizionale. Nei casi metastatici la TC e la RM integrate con l’imaging funzionale (68-Ga-PET/TC) sono fondamentali per la corretta stadiazione. In particolare, per le neoplasie rettali, la RM ha dimostrato di essere molto utile nella stadiazione locale della neoplasia e fondamentale per pianificare la strategia chirurgica (6). Di grande importanza è anche la l’eco-endoscopia, che permette di dettagliare in maniera precisa l’infiltrazione della mucosa (7).
Terapia chirurgica
NEN colon. Lesioni < 2 cm possono essere asportate endoscopicamente mediante polipectomia o resezione mucosa endoscopica (EMR), mentre le lesioni > 2 cm e/o con istologia aggressiva (G3) e/o con riscontro istologico di invasione della muscolaris mucosae richiedono un intervento più radicale di colectomia.
NEN retto. Nelle neoplasie < 1 cm la resezione endoscopica è in genere curativa, mentre per quelle > 2 cm l’intervento di riferimento è la resezione anteriore bassa. Per le neoplasie di dimensione intermedia (1-2 cm), è suggerito un atteggiamento più aggressivo in presenza di cellule con elevato indice replicativo (G3), invasione della muscolaris mucosae e presenza di linfoadenopatie.
Terapia farmacologica
Gli analoghi della somatostatina così come la terapia radiometabolica possono avere un ruolo, anche se sono molto ridotti i dati circa la loro efficacia anti-proliferativa in questi specifici tipi di tumori.
La chemioterapia a base di streptozotocina + 5-FU o doxorubicina è stata utilizzata per molti anni, pur con tassi di risposta piuttosto bassi (intorno al 25%). Un recente studio italiano ha dimostrato un beneficio clinico (risposta parziale + stabilizzazione di malattia) in circa l’80% dei casi con l’associazione capecitabina + oxaliplatino (8).
Follow-up
Le lesioni < 1 cm senza ulteriori fattori di rischio non necessitano di follow-up dopo l’asportazione, mentre per lesioni > 2 cm (G1-G2) dovrebbe essere programmata colonscopia/rettosigmoidoscopia annuale insieme a TC o RM pelvi. In presenza di neoplasie ad andamento più aggressivo (G3), dovrebbe essere programmato controllo endoscopico e radiologico ogni 4-6 mesi (9).
Bibliografia
- Yao JC, Hassan M, Phan, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008, 26: 3063-72.
- García-Carbonero R, Capdevila J, Crespo-Herrero G, et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETS): results from the National Cancer Registry of Spain (RGETNE). Ann Oncol 2010, 21: 1794–803.
- Ito T, Sasano H, Tanaka M, et al. Epidemiological study of gastroenteropancreatic tumors in Japan. J Gastroenterol 2010, 45: 234–43.
- Modlin IM, Lye KD, Kidd MA. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97: 934-59.
- Shimizu T, Tanaka S, Haruma K, et al. Growth characteristic of rectal carcinoid tumors. Oncology 2000, 59: 229-37.
- Taylor FG, et al. Preoperative high resolution magnetic resonance imaging can identify good prognosis stage I, II, II rectal cancer best managed by surgery alone: a prospective, multicenter, European study. Ann Surg 2011, 253: 711–9.
- Matsumoto T, et al. Endoscopic ultrasonography in rectal carcinoid tumors: contribution to selection of therapy. Gastrointest Endosc 1991, 37: 539–42.
- Bajetta E, et al. Are capecitabine and oxaliplatin (XELOX) suitable treatment for progressing low-grade and high-grade neuroendocrine tumors? Cancer Chemother Pharmacol 2007, 59: 637-42.
- Caplin M, et al. ENETS consensus guidelines for the management of patients with digestive neuroendocrine neoplasms: colorectal neuroendocrine neoplasms. Neuroendocrinology 2012, 95: 88-97.
NET toracici
Sara Pusceddu
Oncologia Medica 1, ENET Center of Excellence, Fondazione IRCCS Istituto Tumori Milano
CLASSIFICAZIONE, EPIDEMIOLOGIA E CLINICA
Le neoplasie neuroendocrine (NET) del polmone sono un gruppo eterogeneo di tumori, con differenti aspetti morfologici e diversi livelli di aggressività clinica. La definizione patologica secondo i criteri diagnostici dell’Organizzazione Mondiale della Sanità (OMS) rappresenta il “gold standard” nella diagnostica dei NET polmonari. La terminologia da usare per descrivere i NET polmonari è quella contenuta nella classificazione OMS, edizione 2004, che identifica quattro varianti morfologiche (tabella) (1).
| Classificazione NET polmonari secondo OMS 2004 |
| Carcinoide tipico (CT) Carcinoide atipico (CA) Carcinoma NE a grandi cellule (large cell neuroendocrine carcinoma, LCNEC) Carcinoma a piccole cellule (small cell neuroendocrine carcinoma, SCLC) |
Dati epidemiologici indicano un aumento dell’incidenza di CT e CA, dovuto sia ad una migliorata consapevolezza diagnostica che all’uso sistematico delle colorazioni immuno-istochimiche, mentre i SCLC appaiono in declino, verosimilmente per la progressiva riduzione dell’abitudine tabagica (2). Dati epidemiologici sui LCNEC scarseggiano, ma essi appaiono sempre più frequentemente diagnosticati.
I NET polmonari rappresentano l’1–2% di tutti i tumori maligni polmonari e il 10–30% di tutte le neoplasie neuroendocrine. Spesso la diagnosi è occasionale, ma quando la malattia si manifesta in forma sintomatica, i sintomi d’esordio più frequenti sono rappresentati dall’ostruzione delle vie aeree, quindi tosse, dispnea, emottisi e stridore (3).
Queste neoplasie sono raramente funzionanti (1-5% dei casi) e quando la sindrome è manifesta, essa è maggiormente correlata alla secrezione ectopica di ACTH piuttosto che di serotonina. Nel 5-15% dei casi NET bronchiali e timici possono far parte di sindromi endocrine multiple (MEN-1) (4).
Dati epidemiologici, genetici, patologici e clinici indicano che i CT sono tumori maligni di basso grado, i CA tumori maligni di grado intermedio e LCNEC/SCLC tumori maligni di alto grado, con caratteristiche prognostiche differenti. CT e CA rappresentano un gruppo di tumori ben differenziati (WD-NET) a comportamento clinico indolente, in contrapposizione a LCNEC e SCLC che sono carcinomi scarsamente differenziati (PD-NEC) caratterizzati da andamento clinico aggressivo e rapidamente evolutivo. Nell’attuale classificazione non è accettato un sistema di “grading” a tre livelli (G1, G2, G3), che è invece “ il fondamento” della classificazione morfologica OMS dei GEP-NET sulla scorta di Ki-67 e conta mitotica.
Il ruolo del KI-67 non è ancora ben codificato nei NET polmonari (4). Tuttavia, i suoi ambiti d’utilizzo possono essere esemplificati come segue:
- utilità nel distinguere CT e CA da NEC scarsamente differenziati, in particolare SCLC, nel materiale diagnostico limitato (citologia e biopsie) (5);
- possibilità di usare il Ki-67 come criterio prognostico (in letteratura sono stati proposti vari “cut-off”) nell’ambito dei carcinoidi, anche con valore indipendente in analisi multivariata, mentre non vi sono dati in tale senso nei NEC scarsamente differenziati.
TRATTAMENTO
Analoghi della somatostatina (SSA)
Gli SSA producono un miglioramento della sintomatologia clinica in oltre il 60% dei casi, stabilizzazione della crescita tumorale nel 30-50% e solo in rari casi determinano una regressione parziale del tumore. In generale, le linee guida internazionali raccomandano l’uso degli SSA nei NET sindromici e nei NET a basso grado di malignità non funzionanti se evolutivi (6).
L’evidenza di un effetto anti-proliferativo degli SSA nei NET toracici deriva essenzialmente da studi retrospettivi su casistiche eterogenee di pazienti e da studi clinici prospettici condotti sui GEP-NET (7-8). Unico studio prospettico dedicato ai NET polmonari è lo studio internazionale LUNA, di fase II. Tale studio, prospettico, multicentrico, randomizzato, valuterà l’attività di Pasireotide (SSA che si lega a 4 dei 5 sottotipi recettoriali della somatostatina) ed Everolimus da soli (single arm) oppure in combinazione (Everolimus + Pasireotide) nei NET toracici ben differenziati. Lo studio è al momento in corso di reclutamento e i risultati non sono ancora disponibili (9).Il trattamento adiuvante con SSA nei pazienti con carcinoidi polmonari o timici radicalmente resecati non è stato sufficientemente studiato e non è al momento indicato. A tale scopo sono necessari studi randomizzati specificamente disegnati e con adeguato potere statistico.
Everolimus
Lo studio Italiano di Righi et al, ha analizzato l’espressione in vitro di mTOR fosforilato (p-mTOR) e del suo downstream p70-S6K (p-S6K) e 4EBP1 (p-4EBP1) in un’ampia serie di 218 NET polmonari resecati (24 CT, 73 CA, 60 LCNEC e 61 SCLC). L’analisi ha evidenziato elevati livelli immunoistochimici di p-mTOR e di p-S6K (indicatori di attivazione della via di segnale di m-TOR) nelle forme a basso grado (CT e CA) rispetto alle forme ad alto grado. Inoltre p-mTOR risultava positivamente associato all’espressione dei recettori della somatostatina (10). In analogia, studi pre-clinici hanno confermato l’attività anti-proliferativa di Everolimus su linee cellulari di carcinoidi polmonari (11).
Contrariamente all’esperienza pre-clinica, l’esperienza clinica sull’uso di Everolimus nei NET toracici è molto eterogenea, per assenza di dati relativi a studi retrospettivi o prospettici dedicati a questo sottogruppo di pazienti; inoltre, i dati sono di difficile valutazione nel limitato numero di pazienti arruolati in studi clinici controllati. Lo studio di fase III, multicentrico, internazionale (RADIANT-2), che ha confrontato Everolimus più Octreotide LAR versus placebo più Octreotide LAR in 429 pazienti affetti da NET ben differenziato, funzionante, ha arruolato 44 NET di origine polmonare. Non essendo stata prevista dall’inizio una stratificazione per sede della neoplasia primitiva, la distribuzione dei pazienti è risultata sbilanciata nei due bracci di trattamento (11 pazienti nel braccio placebo e 33 pazienti nel braccio everolimus). L’analisi per sottogruppo, effettuata dopo la pubblicazione dei dati dello studio RADIANT-2, ha confermato, in analogia ai dati complessivi dello studio, un beneficio clinico nei NET toracici trattati con Everolimus rispetto al placebo (miglioramento di 2.4 volte rispetto a placebo della sopravvivenza libera da malattia – mPFS –, da 5.6 a 13.6 mesi) seppur in assenza di una significatività statistica (12).Seppur più limitata, una differente popolazione di NET toracici è stata recentemente valutata nello studio di fase II, RAMSETE: 22 NET toracici non funzionanti a basso grado di malignità, in progressione, sono stati arruolati con l’intento di valutare l’attività di Everolimus in monoterapia, dimostrando un controllo della crescita tumorale nel 60% dei casi (13).
Sulla scorta di tali evidenze, nel 2011 è stato disegnato lo studio RADIANT-4, prospettico, randomizzato, placebo-controllato, in doppio cieco, di fase III, includente pazienti affetti da NET non funzionante sia GEP sia polmonare. L’arruolamento dello studio si è concluso nel settembre 2013 e i risultati non sono ancora disponibili (14).
L’unico studio prospettico specifico per carcinoidi polmonari e timici è lo studio LUNA, già sopra citato (9).
Chemioterapia nei NET toracici a basso grado
La chemioterapia sistemica è riservata ai casi di malattia localmente avanzata o a distanza. Considerata la rarità clinica e la mancanza di adeguati livelli di evidenza, a causa dell’assenza di studi prospettici e retrospettivi sui NET polmonari, al momento non esiste uno schema chemioterapico standard di riferimento da raccomandare nella pratica clinica. Inoltre, data la loro bassa attività proliferativa, i carcinoidi sono da ritenersi in generale neoplasie chemio-resistenti (15).
Le terapie basate su un singolo agente chemioterapico hanno dimostrato tassi di risposta obiettiva generalmente non > 20%, pertanto si tende a riservare una mono-chemioterapia a pazienti pre-trattati o a pazienti con scarso performance status o comorbilità di rilievo. I farmaci più frequentemente utilizzati in mono-chemioterapia sono stati 5-fluoro-uracile, cisplatino, carboplatino, irinotecan, temozolomide, gemcitabina, etoposide, doxorubicina, streptozotocina, dacarbazina, paclitaxel, docetaxel, pemetrexed.
I regimi di poli-chemioterapia hanno mostrato maggiore attività, come emerge da analisi retrospettive e studi di fase II e pertanto sono da preferire, in assenza di controindicazioni. La poli-chemioterapia è in grado di determinare risposte parziali (RP) radiologiche nel 5-10%, stabilità di malattia (SD) nel 30-50%, risposte sintomatiche nel 40-60% dei casi. Tuttavia, non dobbiamo dimenticare che tali risultati sono estrapolati da studi condotti su pazienti affetti da NET di qualsiasi sede anatomica e in cui sono inseriti anche i pazienti con carcinoidi bronchiali e timici. Questo ovviamente ci porta a dover ridurre i livelli di evidenza degli studi, anche per studi ben condotti e con bassa probabilità di bias.
Tra alchilanti e anti-metaboliti la temozolomide è il farmaco più innovativo e meno tossico. La somministrazione orale e la possibilità di essere associata ad altri citostatici rendono temozolomide preferibile a dacarbazina, con cui condivide alcuni metaboliti. La temozolomide è inoltre in grado di attraversare la barriera emato-encefalica e può essere utilizzata per lunghi periodi di tempo. Schemi contenenti temozolomide possono essere di beneficio per il trattamento dei carcinoidi in fase avanzata. In uno studio retrospettivo condotto su 36 pazienti affetti da NET, di cui 7 carcinoidi timici e 13 carcinoidi bronchiali, la somministrazione di temozolomide da sola ha determinato RP nel 14% dei pazienti e SD nel 53%. La mPFS è stata di 7 mesi e la tossicità più rilevante è stata ematologica (piastrinopenia di grado 3 e 4) nel 14% dei casi (16).
Un secondo studio retrospettivo ha valutato l’attività di temozolomide in 31 pazienti affetti da carcinoidi bronchiali metastatici, riportando RP nel 14% dei casi e SD nel 52% dei casi. La tossicità di grado 3 e 4 maggiormente riscontrata è stata, come atteso, la trombocitopenia (17).
Recenti evidenze sembrerebbero identificare la metil-guanin-metil-transferasi (MGMT) nelle cellule tumorali, come fattore predittivo di risposta al trattamento con temozolomide. MGMT è l’enzima responsabile della riparazione del DNA dai danni causati da agenti alchilanti come la temozolomide: alti livelli intra-cellulari di enzima ostacolerebbero l’attività del farmaco. Da un’analisi retrospettiva su 97 pazienti con NET avanzato (pancreas, intestino e polmone) trattati con temozolomide è emerso che la metilazione genica di MGMT (che abbassa i livelli di enzima intra-cellulare) è più frequente nelle neoplasie pancreatiche rispetto ai carcinoidi, così come il tasso di RP al trattamento con temozolomide (34% nei NET pancreatici vs 2% nei carcinoidi) (18).Al momento l’utilizzo di temozolomide non è vincolato dalla determinazione di MGMT, poiché è necessaria una maggiore validazione di questo test e una divulgazione della metodologia per la sua misurazione, oggi prerogativa di pochi centri.
Derivati del platino
Nelle casistiche più datate i carcinoidi bronchiali venivano trattati con lo stesso schema utilizzato per i NET polmonari più aggressivi, in analogia allo schema standard utilizzato nel microcitoma polmonare (Cisplatino-Etoposide).
In realtà i carcinoidi, caratterizzati da una più bassa attività replicativa, sono risultati meno sensibili all'azione di tale regime (19). Inoltre, il cisplatino non era mai stato sistematicamente testato per il trattamento di prima linea in un gruppo omogeneo di NET ben differenziati. Da segnalare, tuttavia, esperienze più recenti, seppur su casistiche limitate, sull’utilizzo di schemi di trattamento contenenti platino. In particolare, nel 2010 sono stati resi noti i risultati di uno studio di fase 2, in cui 98 pazienti affetti da NET ben differenziati, avanzati (8 carcinoidi polmonari) sono stati trattati con un regime polichemioterapico contenente cisplatino, 5-fluoro-uracile e streptozotocina (20). Sono state ottenute RP nel 25% dei casi extra-pancreatici e nel 25% dei carcinoidi polmonari, con SD globalmente nel 51% dei casi. mPFS e sopravvivenza complessiva sono state rispettivamente di 9.1 mesi e 31.5 mesi.
Nel 2006 un gruppo di ricercatori Italiani ha valutato l’attività del regime XELOX (capecitabina + oxaliplatino), nei pazienti affetti da NET a basso e alto grado di malignità sia GEP che toracici. Tra i 27 pazienti valutati, 5 erano affetti da carcinoide polmonare. L’analisi dei risultati ha evidenziato RP nel 27.5% dei casi e SD nel 35% dei casi, con mPFS di 18 mesi. In particolare, gli autori segnalavano un elevato tasso di risposte obiettive e stabilizzazioni di malattia nell’80% dei pazienti affetti da carcinoidi polmonari, riportando viceversa una scarsa risposta clinica nei pazienti affetti dalle forme scarsamente differenziate(21).
Concludendo, in assenza di studi clinici, prospettici, randomizzati, disegnati ad hoc per i NET del distretto toracico, nella pratica quotidiana dobbiamo prediligere il regime chemioterapico esclusivamente sulla base di un ragionamento clinico, tenendo conto di caratteristiche del paziente ed istologia, considerando sempre come prerogativa valida, quando fattibile, l’inserimento del paziente nell’ambito di un trial clinico.
BIBLIOGRAFIA
- Travis W, Brambilla E, Muller-Hermelink H, et al. Tumours of the lung, pleura, thymus and heart. Lyon: IARC Press; 2004.
- Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003, 97: 934-59.
- Rekhtman N. Neuroendocrine tumors of the lung: an update. Arch Pathol Lab Med 2010, 134: 1628-38.
- Phan AT, Oberg K, Choi J, et al. NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors: well-differentiated neuroendocrine tumors of the thorax (includes lung and thymus). Pancreas 2010, 39: 784-98.
- Pelosi G, Rodriguez J, Viale G, et al. Typical and atypical pulmonary carcinoid tumor overdiagnosed as small-cell carcinoma on biopsy specimens: a major pitfall in the management of lung cancer patients. Am J Surg Pathol 2005, 29: 179-87.
- Modlin IM, Pavel M, Kidd M, et al. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 2010, 31: 169–88.
- Rinke A, Müller HH, Schade-Brittinger C, et al.Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009, 27: 4656-63.
- Caplin ME, Pavel M, Ćwikła JB, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014, 371: 224-33.
- Multicenter 3-arm trial to evaluate the efficacy and safety of pasireotide LAR or everolimus alone or in combination in patients with well differentiated neuroendocrine carcinoma of the lung and thymus - LUNA Trial. clinicaltrials.gov NCT01563354.
- Righi L, Volante M, Tavaglione M, et al. Mammalian target of rapamycin (mTOR) signaling activation patterns in neuroendocrine tumors of the lung. Endocr Relat Cancer 2010, 17: 977-87.
- Zatelli MC, Minoia M, Martini C, et al. Everolimus as a new potential antiproliferative agent in aggressive human bronchial carcinoids. Endocr Relat Cancer 2011, 17: 719-29.
- Fazio N, Granberg D, Grossman A, et al. Everolimus plus octreotide LAR in patients with advanced lung neuroendocrine tumors: analysis of the phase III, randomized, placebo-controlled RADIANT-2 study. Chest 2013, 143: 955-62.
- Pavel M, Wiedenmann P, Capdevila J, et al. RAMSETE: a single-arm, multicenter, single stage phase III trial of RAD001 (everolimus) in advanced and metastatic silent neuro-endocrine tumours in Europe: analysis by tumor origin. Ann Oncol 2012, 23 suppl 9: 11560.
- Everolimus plus best supportive care vs placebo plus best supportive care in the treatment of patients with advanced neuroendocrine tumors (GI or lung origin) (RADIANT-4). Clinicatrials.gov NCT01524783.
- Lim E, Goldstraw P, Nicholson G, et al. Proceedings of the IASLC international workshop on advances in pulmonary neuroendocrine tumors 2007. J Thorac Oncol 2008, 3: 1194–201.
- Eklebad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res 2007, 13: 2986-91.
- Crona J, Fanola I, Lindholm DP, et al. Effect of temozolomide in patients with metastatic bronchial carcinoids. Neuroendocrinology 2013, 98: 151-5.
- Kulke MH, Hornick JL, Frauenhoffer C, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res 2009, 15: 338-45.
- Moertel CG, Kvols LK, O’Connell MJ, et al. Treatment of neuroendocrine carcinomas with combined etoposide and cisplatin. Evidence of major therapeutic activity in anaplastic variance of these neoplasm. Cancer 1991, 68: 227–32.
- Turner NC, Strauss SJ, Sarker D, et al. Chemotherapy with 5-fluorouracil, cisplatin and streptozotocin for neuroendocrine tumours. Br J Cancer 2010, 10: 1106-12.
- Bajetta E, Catena L, Procopio G, et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother Pharmacol 2007, 59: 637-42.
NET rari
Fernando Cirillo
Dipartimento di Chirurgia Generale, Unità di Chirurgia Generale, Gruppo Tumori Ormonali Rari, AO Istituti Ospitalieri, Cremona
I Tumori Neuroendocrini (NET) rari rappresentano un piccolo gruppo di neoplasie di osservazione quasi aneddotica se confrontata con quella dei più comuni NET. Al gruppo appartengono principalmente i carcinomi epiteliali con differenziazione NE e i tumori con fenotipo NE – che chiameremo “puri” – cresciuti in distretti considerati insoliti per questo tumore; completano il gruppo i paragangliomi e le associazioni fra NET ed altri tumori rari.
Dal momento che le segnalazioni in letteratura sono per lo più aneddotiche, i riferimenti alla diagnosi e alla terapia mancano di un consenso tipico delle più ampie casistiche. In linea del tutto generale, la diagnosi di questi tumori è del tutto casuale: l’imaging radiologico, nella maggior parte dei casi, provvede a descrivere lesioni non-funzionanti nonostante i caratteri ormonali di questi tumori. Il trattamento standard è chirurgico nelle forme primitive; la chemioterapia è di necessità nelle forme avanzate a differenziazione NE per le caratteristiche biologiche che li avvicinano alla controparte epiteliale. L’uso aneddotico di altre molecole (analoghi della somatostatina, inibitori di mTOR, inibitori di VEGFR, terapia radio-recettoriale) non ha offerto risposte definitive. Poco descritto in letteratura il ruolo del follow-up.
CARCINOMI CON DIFFERENZIAZIONE NE
La classificazione WHO relativa ai tumori del sistema digestivo pubblicata nel 2010 (1) descrive un gruppo di tumori misti, dove la componente esocrina ed endocrina deve essere rappresentata arbitrariamente con una quota pari al 30%; quando la componente endocrina sia presente con una quota < 30%, è possibile utilizzare il termine di carcinoma epiteliale a differenziazione NE. La metodica immunoistochimica utilizzata e il diverso grado di sensibilità possono influenzare la diagnosi sul preparato istologico (2).
La presenza di una differenziazione in senso NE può avere un riflesso prognostico diverso a seconda dei distretti interessati. Ad esempio, tale condizione non sembra influenzare la prognosi per il carcinoma mammario e del colon-retto; diverso è il caso del carcinoma gastrico, prostatico e appendicolare, dove tale differenziazione si associa ad una prognosi sfavorevole; infine, per il carcinoma pancreatico la prognosi è fortemente influenzata dalla componente esocrina (3). È stata anche segnalata una prognosi peggiore nei tumori esofago-gastrici e del retto con differenziazione NE radio-chemio trattati (3). Nel distretto respiratorio esiste un’area di sovrapposizione, rappresentata dai tumori squamosi (non oat cell) con differenziazione NE, che costituisce una quota significativa pari al 15-20% dei casi (4). Tale condizione non sembra influenzare in maniera significativa la prognosi.
In tutte le situazioni descritte sintomi e segni clinici non sono per nulla differenti rispetto alla controparte epiteliale. Il ruolo del patologo in tutti questi casi è cruciale, perché le informazioni destinate al clinico, integrate con la diagnostica PET (68Ga e 18F-FDG), saranno fondamentali per la strategia terapeutica.
TUMORI CON FENOTIPO NE (“puri”)
Per questi tumori la letteratura è relativamente scarsa. Tale carenza può essere spiegata per la loro frequente sintomaticità e per l’estrema distribuzione in più distretti dell’organismo. I NET “puri” presentano incidenza < 1% e corrispondono circa al 10-15% di tutti i NET (5). In relazione alla sede, possono essere suddivisi in: testa/collo, timo, mammella, gonadi, rene, vescica, prostata e pancreas.
Testa/collo – Laringe. I tumori NE del laringe coprono circa l’1% di tutte le neoplasie laringee e insorgono più di frequente in maschi anziani fumatori. Sintomi caratteristici sono dolore, disfagia, raucedine e dispnea. Si tratta di una neoplasia rara e aggressiva, con una sopravvivenza spesso non correlata alla radicalità della chirurgia, dove le metastasi a distanza rappresentano la causa di decesso (6). Sono stati pubblicati 127 casi di carcinoide atipico del laringe, argirofilo nel 97% dei casi, con prognosi severa nei pazienti metastatici o con lesione primitiva con diametro > 1 cm (7). L’istotipo a piccole cellule rappresenta una forma inusuale, pari allo 0.5% di tutti i carcinomi laringei. Nel 73% dei casi il decesso è secondario alla malattia metastatica; radio e chemioterapia portano la sopravvivenza al 5% a 5 anni (8). I tumori NE dell’esofago rappresentano lo 0.05% di tutte le neoplasie gastro-esofagee: interessano il terzo inferiore dell’esofago e insorgono di solito nei maschi dopo la 6° decade di vita. Disfagia, dolore, reflusso gastro-esofageo e calo ponderale sono i segni e sintomi più frequenti. La diagnosi è strumentale. Le caratteristiche morfologiche e immunoistochimiche sono quelle caratteristiche dei tumori NE, con espressione di cromogranina A e NSE (5).
Timo. Le neoplasie NE del timo rappresentano una quota < 3% rispetto al numero totale dei NET. Asintomatici al momento della diagnosi in 1/3 dei casi (9), si presentano già metastatici nel 30% dei casi (5). Possono essere a carattere sporadico o nell’ambito di una MEN-1 nel 5% dei casi (5,10). Le forme sporadiche presentano sintomi locali da compressione (tosse stizzosa, sindrome da compressione venosa) o sindromi paraneoplastiche, in particolare la sindrome di Cushing in 1/3 dei casi osservati (11), ma anche la polimiosite e le cardiomiopatie. La diagnosi è strumentale: la RMN descrive questa neoplasia come una massa a contorni policiclici e irregolari e con presenza di calcificazioni. L’elevata aggressività del tumore richiede il provvedimento chirurgico, con prognosi infausta nel 60% dei casi (12,13).
Mammella. Si tratta di un’entità rara, pari al 5% di tutte le neoplasie mammarie (14). La neoplasia esprime i recettori estrogenici, cromogranina A e B, sinaptofisina e NSE. La fascia di età colpita è molto ampia (38-87 anni) (5). L’obiettività mammaria non è diversa da quella del comune carcinoma epiteliale: questi tumori possono essere, infatti, unici, multifocali e bilaterali. L’aspetto macroscopico non è considerato caratteristico. Non si conosce la sindrome da carcinoide neppure in presenza di malattia disseminata. Sono stati invece riportati rari casi di sindrome di Cushing associata (15). I dati di follow-up su 30 casi indicano la tendenza ad un buon andamento della malattia (mediana di sopravvivenza libera da malattia di 48 mesi) (16). Questa neoplasia è stata segnalata in un maschio di 80 anni associata a una componente papillare con localizzazioni ascellari (17). La terapia è chirurgica in tutte le forme primitive descritte.
Gonadi. Nel sesso femminile, il carcinoma NE ovarico, che assomma a circa il 2% di tutte le neoplasie ginecologiche (18), si presenta di solito come localizzazione monolaterale, unica, non invasiva, talvolta associata a elementi teratomatosi. È presente la sindrome da carcinoide nel 30% dei casi (5): di questa, in una casistica di 329 casi è riportata una diversa incidenza per il tipo “insulare” (39%) rispetto a quello trabecolare (8%) (19). La prognosi è favorevole nella malattia localizzata. Nelle localizzazioni secondarie da NET digestivo il comportamento è invece più aggressivo (5). In questi casi il trattamento medico prevede l’uso di analoghi della somatostatina, IFNα, inibitori di mTOR, embolizzazione epatica e terapia radio-recettoriale (18). È riportato il caso di un carcinoma NE a grandi cellule del corpo dell’utero in una paziente di 52 anni (20) e il caso di un carcinoma NE della cervice uterina. Per quest’ultimo istotipo, le metastasi loco-regionali sono precoci nel 68% dei casi con recidiva nella metà dei casi (21). Nel maschio, il carcinoma NE del testicolo colpisce un’ampia fascia di età, fino oltre gli 80 anni. Insorge prevalentemente nel testicolo sinistro come massa localizzata che determina turgore dello scroto, dolore spontaneo e rischio di torsione (5). Sono segnalati 57 casi con caratteristiche prevalentemente argirofile e l’associazione col teratoma in 43 casi. Un maggior potenziale metastatico è correlato con le dimensioni del tumore e con la sindrome da carcinoide (22). La diagnosi è clinica e strumentale, la terapia chirurgica (5). Raro è il carcinoide “puro” della prostata, mentre sono di numero maggiore le segnalazioni di una differenziazione NE come dimostrato dall’espressione dei marcatori tipici di malattia. Dal momento che la terapia ormonale ablativa non è in grado di controllare la crescita neoplastica della popolazione cellulare con fenotipo NE, i cloni cellulari con recettori androgeno – negativi proliferano con modalità del tutto autonome (23). Ciò si correla con una prognosi peggiore (23, 24) che implica diverse soluzioni terapeutiche. Infine, è stato segnalato il caso di un carcinoide dell’epididimo sottoposto ad orchiectomia bilaterale (25).
Rene. Ancora meno frequente è la localizzazione renale, di cui sono segnalati 14 casi (26).
Vescica. Sono stati segnalati 1 caso con istotipo a piccole cellule (0.35-0.70% di tutte le neoplasie vescicali) (27) e 1 caso con istotipo a grandi cellule (28). La macroematuria rappresenta l’insorgenza clinica della malattia. La terapia è combinata.
Pancreas. Soga ha segnalato 156 casi di carcinoide “puro” del pancreas, pari all’1.4% di tutti i NET registrati nel Niigata Registry for Gut-Pancreatic Endocrinomas, di cui 144 tipici e 12 atipici. Essi presentano un elevato trend metastatico (66.7%), hanno un diametro medio (68.6 mm) e una incidenza della sindrome da carcinoide (23.3%) superiori rispetto a tutti gli altri NET (29).
Il paraganglioma deriva dal tessuto cromaffine extra-surrenalico e può interessare sia il sistema nervoso simpatico che parasimpatico. Si tratta di un tumore con aspetti NE che, quando funzionante, può determinare sintomi tipici del feocromocitoma, comprese le severe complicanze cardio-vascolari. In letteratura sono segnalati casi sporadici di paraganglioma a carico di differenti distretti. Sono 28 i casi riportati a carico del collo (bulbo carotideo, nervo vago, vena giugulare, laringe) (30), 9 a carico della tiroide (31), 1 in sede intra-pericardica (32), 80 casi di paraganglioma gangliocitico pancreatico (33), 6 casi a carico della prostata (34), infine 1 caso localizzato nella regione pelvica (35). La terapia è chirurgica nelle fasi iniziali. Va considerato il trattamento dell’ipertensione arteriosa, con le associazioni α-litico e β-bloccante nelle forme funzionanti e la chemioterapia negli stadi avanzati della malattia. In un caso osservato, il trattamento con sunitinib ha consentito una discreta risposta obiettiva in un giovane paziente già sottoposto a numerosi e differenti trattamenti (36).
NET E ASSOCIAZIONE CON ALTRI TUMORI RARI
In casi del tutto eccezionali si possono osservare associazioni fra NET e altre neoplasie rare, prevalentemente epiteliali. Di queste, sono segnalati 4 casi con una percentuale di osservazione pari all’1.3% (37). L’asportazione della lesione è lo standard terapeutico. Il follow-up deve considerare tempi stretti. In casi selezionati la terapia è farmacologica.
BIBLIOGRAFIA
- Bosman FT, Carneiro F, Hruban RH, et al. WHO classification of tumours of digestive system. International Agency for Research on Cancer, Lyon 2010: pg 14.
- Papotti M. Tumori a differenziazione neuroendocrina: il punto di vista del patologo. In: Cirillo F (ed), Tumori a differenziazione neuroendocrina: implicazioni diagnostiche e terapeutiche. J Health Sci, MB & CARE, Livorno 2010, anno 8, n° 3. Atti del corso, Cremona 12 febbraio 2010; pgg 6-8.
- Fazio N. La differenziazione neuroendocrina nel distretto digestivo. In: Cirillo F (ed), Tumori a differenziazione neuroendocrina: implicazioni diagnostiche e terapeutiche. J Health Sci, MB & CARE, Livorno 2010, anno 8, n° 3. Atti del corso, Cremona 12 febbraio 2010; pgg 9-10.
- Carnaghi C. La differenziazione neuroendocrina nel distretto respiratorio. In: Cirillo F (ed), Tumori a differenziazione neuroendocrina: implicazioni diagnostiche e terapeutiche. J Health Sci, MB & CARE, Livorno 2010, anno 8, n° 3. Atti del corso, Cremona 12 febbraio 2010; pgg 11-12.
- Grimaldi G. Gli altri distretti: testa-collo, timo, mammella, gonadi. In: Cirillo F (ed), Tumori a differenziazione neuroendocrina: implicazioni diagnostiche e terapeutiche. J Health Sci, MB & CARE, Livorno 2010, anno 8, n° 3. Atti del corso, Cremona 12 febbraio 2010; pgg 13-15.
- Baugh RF, Wolf GT, Lloyd RV, et al. Carcinoid (neuroendocrine carcinoma) of the larynx. Ann Otol Rhinol Laryngol 1987, 96: 315-21.
- Woodruff LM, Senie RT. Atypical carcinoid tumor of the larynx. A critical review of the literature. ORL J Otorhinolaryngol Relat Spec 1991, 53: 194-209.
- Gnepp DR. Small neuroendocrine carcinoma of the larynx. A critical review of the literature. ORL J Otorhinolaryngol Relat Spec 1991, 53: 210-9.
- Moran CA, Suster S. Neuroendocrine carcinomas (carcinoid tumor) of the thymus. A clinicopathologic analysis of 80 case. Am J Clin Pathol 2000, 114: 100-10.
- Zeiger MA, Swartz SE, MacGillivray DC, et al. Thymic carcinoid in association with MEN sindrome. Am Surg 1992, 58: 430-4.
- Sugiura H, Morikawa T, Itoh K, et al. Thymic neuroendocrine carcinoma: a clinicopathologic study in four patients. Ann Thorac Cardiovasc Surg 2000, 6: 304-8.
- Gal AA, Kornstein MJ, Cohen C, et al. Neuroendocrine tumors of the thymus: a clinicopathological and prognostic study. Ann Thorac Surg 2001, 72: 1179-82.
- De Montpreville VT, Macchiarini P, Dulmet E. Thymic neuroendocrine carcinoma (carcinoid): a clinicopathologic study of fourteen cases. J Thorac Cardiovasc Surg 1996, 111: 134-41.
- Azzopardi JG, Muretto P, Goddeeris P, et al. “Carcinoid” tumours of the breast: the morphological spectrum of argyrophilic carcinomas. Histopathology 1982, 6: 549-69.
- Cohele SD, Tschen JA, Smith FE, et al. ACTH-secreting carcinoma of the breast. Cancer 1979, 43: 2370-6.
- Colombo L, Riva C, Fabbri A, et al. I carcinomi neuroendocrini della mammella. Congresso Società Italiana di Chirurgia Generale, Roma ottobre 1996. Pozzi Editore: pg 82-95.
- Lingappa HA, Indushekar V, Chamarthy NP, et al. Primary neuroendocrine carcinoma of male breast: a cytologically diagnosed rare entity. J Cytol 2014, 31: 105-7.
- Reed NS, Gomez-Garcia E, Gallardo-Rincon D, et al. Gynecologic Cancer InterGroup (GCIG) consensus review fo carcinoid tumors of the ovary. Int J Ginecol Cancer 2014, 24 (9 suppl 3): S35-41.
- Soga J, Osaka M, Yakuwa Y. Carcinoids of the ovary: an analysis of 329 reported cases. J Exp Clin Cancer Res 2000, 19: 271-80.
- Erhan Y, Dikmen Y, Yucebilgin MS, et al. Large cell neuroendocrine carcinoma of the uterine corpus metastatic to brain and lung: case report and review of the literature. Eur J Gynaecol Oncol 2004, 25: 109-12.
- Trinh XB, Bogers JJ, Van Marck EA, et al. Treatment policy of neuroendocrine small cell cancer of the cervix. Eur J Gynaecol Oncol 2004, 25: 40-4.
- Zavala-Pompa A, Ro JJ, El-Naggar A, et al. Primary carcinoid tumor of testis. Immunohistochemical, ultrastructural, and DNA flow cytometric study of three cases with a review of the literature.Cancer 1993, 72: 1726-32.
- Abrahamsson PA. Neuroendocrine differentiation in prostatic carcinoma. Prostate 1999, 39: 135-48.
- McWilliam LJ, Manson C, George NJ. Neuroendocrine differentiation and prognosis in prostatic adenocarcinoma. Br J Urol 1997, 80: 287-90.
- Zeng l, Xia T, Kong X, et al. Primary carcinoid tumor of the epididymis. Chinese Med J 2001, 114: 544-5.
- Sahin A, Demirbas M, Ozen H, et al. Primary carcinoid of the kidney. Scand J Urol Nephrol 1996, 30: 325-7.
- Aragon-Tovar AR, Pineda-Rodriguez ME, Puente-Gallegos FE, et al. Neuroendocrine carcinoma of the urinary bladder. A case report. Cir Cir 2014, 82: 338-43.
- Dundr P, Pesl M, Povysil C, et al. Large cell neuroendocrine carcinoma of the urinary bladder with lymphoepithelioma-like features. Pathol Res Pract 2003, 199: 559-63.
- Soga J. Carcinoid of the pancreas. Cancer 2005, 104: 1180-7.
- Bishop GB jr, Urist MM, el Gammal T, et al. Paragangliomas of the neck. Arch Surg 1992, 127: 1441-5.
- Brownlee RE, Shockley WW. Thyroid paraganglioma. Ann Otol Rhinol Laryngol 1992, 101: 293-9.
- Hamilton BH, Francis IR, Gross BH, et al. Intrapericardial paragangliomas (pheochromocytomas): imaging features. AIR Am J Roentgenol 1997, 168: 109-13.
- Tomic S, Warner T. Pancreatic somatostatin-secreting gangliocytic paraganglioma with lymph node metastases. Am J Gastroenterol 1996, 91: 607-8.
- Hasselager T, Horn T, Rasmussen F. Paraganglioma of the prostate. A case report and review of the literature. Scand J Urol Nephrol 1997, 31: 501-3.
- Taue R, Takigawa H, Sinotou K, et al. A case of pelvic malignant paraganglioma. Int J Urol 2001, 8: 715-8.
- Cirillo F. Metastatic paraganglioma and treatment with sunitinib: a case report. Tumori 2010, 96: 1022-7.
- Cirillo F. Neuroendocrine tumors and their association with rare tumors: observation of 4 case. Eur Rev Med Pharmacol Sci 2010, 14: 577-88.
Carcinoma a cellule di Merkel
Fernando Cirillo
Dipartimento di Chirurgia Generale, Unità di Chirurgia Generale, Gruppo Tumori Ormonali Rari, AO Istituti Ospitalieri, Cremona
DEFINIZIONE ED EPIDEMIOLOGIA
Il carcinoma a cellule di Merkel (MCC) è una neoplasia della cute rara e aggressiva, caratterizzata da frequenti recidive locali, rapida progressione della malattia, scarsa qualità di vita e breve sopravvivenza.
L’origine di questo tumore non è stata provata in modo definitivo e oggi si ritiene che la cellula di Merkel possa derivare da una cellula epiteliale totipotente, in grado di differenziarsi sia in senso neuroendocrino, sia come cheratinocita. A sostegno di questa teoria è la presenza di cellule transizionali simili sia ai cheratinociti che alle cellule di Merkel (1).
Recentemente è stato isolato un polyomavirus presente nell’80% dei MCC con una forte correlazione fra la presenza del virus e stati di alterata risposta immunitaria cellulo-mediata (2).
La reale prevalenza di MCC è sconosciuta. Esso colpisce più frequentemente (3/4 dei casi) pazienti anziani ultra60enni (range 7-95) soprattutto donne (F/M = 3/1). È più comune nelle popolazioni caucasiche, ma occasionalmente presente anche nei negri e nei polinesiani. È stata segnalata un’alta incidenza nei trapiantati, con caratteristiche di maggiore aggressività probabilmente secondaria ad alterazioni del sistema immunocompetente, in un paziente HIV positivo e in due casi di artrite reumatoide (4).
CLINICA, LOCALIZZAZIONE E DIAGNOSI
Le dimensioni della neoplasia possono essere del tutto varie, fino a 15 cm di diametro, con una media alla presentazione di circa 3 cm.
La sede più comune del tumore è la cute della regione testa-collo (50% dei casi), il 40% dei casi interessa le estremità e il rimanente 10% tronco e mucose. Sono stati anche riportati casi di sedi multiple della malattia.
La clinica per MCC permette solo una diagnosi presuntiva; per tale motivo la letteratura anglosassone ha coniato l’acronimo AEIOU (Asymptomatic/lack of tenderness, Expanding rapidly, Immune suppression, Older than age 50, and UV-exposed site on a person with fair skin)(3) per venire in aiuto alla diagnosi. Infatti, se la neoplasia si presenta tipicamente come una lesione solitaria, rilevata o a placca, talvolta peduncolata, di colore rosso-violaceo, a superficie lucida talvolta associata a vicine teleangectasie, nello stadio iniziale della malattia la diagnosi differenziale può risultare poco agevole: MCC può, infatti, essere confuso con basalioma, carcinoma spino-cellulare, granuloma piogenico, cherato-acantoma, melanoma, linfoma cutaneo, metastasi cutanee (da carcinoma a piccole cellule, carcinoma anaplastico, carcinoide, retinoblastoma, sarcoma di Ewing e neuroblastoma).

Merkel all'arto inferiore sinistro (lesione localmente avanzata dopo trattamento incongruo)

Merkel al piede destro

Merkel localmente avanzato da trattamento incongruo (non radicalizzazione, non RT)
I marcatori normalmente espressi da questo tumore sono NSE, cromogranine e sinaptofisina.
La stadiazione della malattia prevede la convenzionale tomografia multistrato (TC) e altre metodiche di immagine ancillari come 18F-FDG-PET-TC, utile come completamento nei pazienti con fattori di rischio più elevati e nel sospetto di recidiva, oppure 68Ga-DOTA-PET/TC per l’elevata espressione di recettori della somatostatina (2). Ciò vale anche per OctreoScan®, che rispetto alla PET con 68Ga presenta minore sensibilità diagnostica (1). Per la stadiazione di MCC viene utilizzata la classificazione AJCC del 2010 (5).
| Classificazione (5) | |||
| T | Tx | Il tumore primitivo non è valutabile | |
| T0 | Nessuna evidenza di tumore primitivo (per esempio il tumore è stato trovato nei linfonodi, ma il tumore principale non è stato trovato) | ||
| Tis | Carcinoma in situ: il tumore è confinato all’epidermide, lo strato più esterno della cute (questo è estremamente raro per il MCC) | ||
| T1 | Diametro tumorale < 2 cm | ||
| T2 | Diametro tumorale 2-5 cm | ||
| T3 | Diametro tumorale > 5 cm | ||
| T4 | Il tumore si è diffuso nei tessuti adiacenti (muscoli, osso, cartilagine) | ||
| N | Nx | I linfonodi regionali non sono valutabili | |
| N0 | I linfonodi regionali non sono interessati dal tumore | pN0: i linfonodi regionali non sono interessati dal tumore all’esame istologico | |
| cN0: i linfonodi regionali non sembrano interessati dal tumore (obiettivamente e agli esami strumentali), ma non sono stati sottoposti a biopsia | |||
| N1 | I linfonodi regionali sono interessati dal tumore all’esame istologico | N1a: ma non lo sembravano agli esami strumentali | |
| N1b: e lo sembravano agli esami strumentali | |||
| N2 | Il tumore si è diffuso verso i linfonodi regionali senza raggiungerli (metastasi in transito) | ||
| M | M0 | Nessuna diffusione metastatica agli organi a distanza | |
| M1a | Il tumore si è diffuso ad altre aree cutanee, ai tessuti sottocutanei, o ai linfonodi a distanza | ||
| M1b | Metastasi polmonari | ||
| M1c | Metastasi extra-polmonari | ||
| Stadiazione | |||
| Stadio | Caratteri | ||
| 0 | Tis, N0, M0 | ||
| I | IA | T1, pN0, M0 | |
| IB | T1, cN0, M0 | ||
| II | IIA | T2 o T3, pN0, M0 | |
| IIB | T2 o T3, cN0, M0 | ||
| IIC | T4, N0, M0 | ||
| III | IIIA | qualsiasi T, N1a, M0 | |
| IIIB | qualsiasi T, N1b o N2, M0 | ||
| IV | qualsiasi T, qualsiasi N, M1 (a, b, o c) | ||
TERAPIA
Trattamento chirurgico
Negli stadi iniziali della malattia (I e II) il trattamento di scelta è rappresentato dall’escissione radicale del tumore primitivo. Allo scopo di evitare la recidiva locale, è raccomandata un’adeguata rimozione della lesione, con margini di almeno 2 cm. La necessità di linfadenectomia elettiva è stata superata con la ricerca del linfonodo sentinella (LS) con tecnica radioisotopica, in grado di localizzare metastasi linfonodali occulte nel 29% dei casi (1). Anche se i pazienti con LS positivo presentano un maggior rischio di recidiva a distanza, sono possibili falsi negativi, con sviluppo di recidiva nel 9.8% dei casi (2).
Radioterapia
La maggior parte degli autori è a favore della radioterapia (RT) adiuvante. La scelta è motivata da una riduzione del rischio di recidiva locale nel I e II stadio della malattia, con presenza di metastasi linfonodali nel 40-73% dei casi e recidiva locale nel 23-60% (1). Questi dati sono suffragati dal programma governativo statunitense Surveillance, Epidemiology, and End Results Program e da quelli del gruppo australiano di Veness (1). Al contrario, la chemioterapia adiuvante non è in grado di ridurre la percentuale di recidiva locale né di migliorare la sopravvivenza. La RT è impiegata come monoterapia nei casi di malattia localmente avanzata in sedi anatomiche critiche con difficile resecabilità.
Chemioterapia
Tradizionalmente viene utilizzata nel setting avanzato, con intento palliativo. I chemioterapici più usati sono etoposide, cisplatino/carboplatino, doxorubicina, dacarbazina, vincristina, ciclofosfamide e metotrexate, sia in monoterapia che in combinazione. Non esistono studi randomizzati di confronto tra vari regimi chemioterapici. In un’analisi retrospettiva su 107 pazienti sono stati osservati tassi di attività con percentuali progressivamente in diminuzione in prima, seconda e terza linea, rispettivamente del 61%, 45%, e del 20%, con una durata di risposta da 3.5 a 15 mesi e sopravvivenza globale a 3 anni pari al 17% nei pazienti metastatici e al 35% in quelli con malattia localmente avanzata. In sostanza, MCC appare una neoplasia chemio-sensibile, ma non curabile con la chemioterapia (2). Lo studio prospettico di fase II TROG (Trans-Tasman Radiation Oncology Group) ha valutato il trattamento sincrono di cisplatino/etoposide associato a RT in pazienti con MCC localmente avanzato e ad alto rischio. Con un follow-up di 48 mesi, la sopravvivenza totale, il controllo loco-regionale e il controllo a distanza sono stati rispettivamente di 76%, 75% e 76%. Questo studio suggerisce che la chemio-radioterapia concomitante può influire positivamente sul controllo loco-regionale e sulla sopravvivenza globale nel MCC localmente avanzato (2).
Altre terapie
L’infiltrazione locale di IFNα-2b, l’utilizzo di Tumor Necrosis Factor (TNF), l’ipertermia associata a basse dosi di RT, la RT associata a TNF-α, IFN-γ e melphalan, l’elettro-chemioterapia e imiquimod associato a RT hanno mostrato aneddoticamente remissioni di malattia, con una sopravvivenza libera da progressione relativamente lunga (2).
I dati di letteratura sono scarsi ed eterogenei per gli analoghi della somatostatina (1,6). Non ci sono evidenze concrete che gli SSA siano attivi nel MCC. Tuttavia, in assenza di alternative terapeutiche, in presenza di gravi comorbilità, in MCC a decorso indolente con espressione di recettori per la somatostatina, gli SSA potrebbero essere presi in considerazione come trattamento palliativo (1).
La co-espressione di c-Kit in un’alta percentuale di MCC suggerisce un ruolo importante degli inibitori di tirosin-chinasi nella trasformazione neoplastica della cellula di Merkel. Su tale base sono state condotte esperienze cliniche con inibitori di c-Kit, che hanno dimostrato evidenze di risposta parziale a imatinib e pazopanib (2).
Terapia radiorecettoriale
Nei pazienti che presentano una positività alla PET/TC con SSA radiomarcati, può essere valutata la possibilità della PRRT con 90Y/177Lu DOTATOC/DOTATATE, di cui sono riportate al momento solo sporadiche esperienze, in base alle quali non è possibile esprimere un giudizio definitivo (2).
CONCLUSIONI
MCC è certamente una neoplasia molto aggressiva e altrettanto sconosciuta. Questi due fattori sono il risultato di numerose osservazioni di recidive locali o di malattia avanzata. Il sito italiano www.neuroendocrini.it dal 2002 ad oggi ha collezionato un considerevole numero di pazienti affetti da MCC con la possibilità di tracciare una mappa dei bisogni, delle differenti risorse culturali e tecnologiche da Regione a Regione (7). Il grande numero di richieste di informazione riguardo a MCC suggerisce che il tumore ha bisogno di attenzione e di expertise, così come oggi succede per il melanoma (8). In un programma di sorveglianza dovremmo di principio inserire il Medico di Medicina Generale, stimolandolo ad una maggiore attenzione alle lesioni della cute e per incoraggiare il paziente ad una precoce asportazione. La tempistica è parte integrante di un percorso appropriato per il management di MCC. Sia il Dermatologo che il Chirurgo e l’Oncologo dovrebbero collaborare per ridurre l’intervallo tra escissione e diagnosi patologica, con lo scopo di rendere il più possibile precoce l’inizio di altre terapie quando necessarie, favorendo una migliore qualità di vita del paziente. Le Linee Guida dovrebbero fornire quelle raccomandazioni chiare e indispensabili per la gestione della neoplasia, facendo il più possibile riferimento alle metodiche e alle terapie accessibili sul nostro territorio, negando i benefici di altre, fornendo anche informazioni sulla spesa sanitaria. Questi potrebbero essere alcuni dei motivi che fanno considerare utile il ruolo dell’informazione sanitaria in rete, soprattutto quando dedicata a patologie poco frequenti e scarsamente conosciute (8).

Strategia per MCC primitivo (sopra) e recidivo (sotto) (1).

BIBLIOGRAFIA
- Cirillo F, Vismarra M, Cafaro I, et al. Merkel cell carcinoma: a retrospective study on 48 cases and review of literature. J Oncol 2012, 2012: 749030.
- AIOM. Linee guida neoplasie neuroendocrine. AIOM/IT.A.NET 2014.
- Heath M, Jaimes N, Lemos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features. J Am Acad Dermatol 2008, 58: 375-81.
- Lima GF, Cirillo F, Buononato M, et al. Clinical experience on eight cases of Merkel cell carcinoma. Tumori 2003, 89: 146-51.
- American Joint Committee. Cancer Staging Manual and Handbook. Merkel Cell Carcinoma. Springer, New York, NY, USA, 2010.
- Cirillo F, Filippini L, Lima GF, et al. Tumore a cellule di Merkel: segnalazione di un caso e trattamento con octreotide. Minerva Chir 1997, 52: 1359-65.
- http://www.neuroendocrini.it/storia.php.
- Cirillo F. Merkel cell carcinoma: need for information and awareness. A case series of 47 patients from an Italian website. Tumori 2014, 100: 504-6.
Patologia neoplastica degli organi endocrini
Neoplasie tiroidee
Neoplasie ipofisarie
Neoplasie paratiroidee
Neoplasie surrenaliche
Neoplasie ovariche
Neoplasie testicolari
Tumori ormono-sensibili e ormono-dipendenti
Overview sui tumori ormono-sensibili e ormono-dipendenti
Valerio Adinolfi, Roberto Baldelli, Antonella Paoloni, Francesca Rota, Laura Rizza, Agnese Barnabei, Marialuisa Appetecchia
UOSD Endocrinologia, Istituto Nazionale Tumori Regina Elena – IRCCS, Roma
INTRODUZIONE
Per la prima volta nel 1963, uno studio pubblicato su Cancer Research (1) individuava tra le cause principali dello sviluppo dei tumori le radiazioni ionizzanti e le sostanze chimiche cancerogene. Tuttavia già in quello studio veniva attribuito un ruolo importante agli ormoni nella crescita di alcune neoplasie di mammella, prostata e utero. Nel 1983, un lavoro (2) concluse che i principali fattori che influenzavano positivamente o negativamente l’incidenza del cancro alla mammella erano le radiazioni ionizzanti, l’annessiectomia bilaterale, l’isterectomia e la terapia ormonale (contraccezione e/o terapia sostitutiva).
Gli estrogeni, attraverso i recettori ERα e ERβ, hanno un ruolo fondamentale nello sviluppo e nella morfogenesi della ghiandola mammaria; entrambi i recettori legano il 17β-estradiolo (E2) con la medesima affinità, ma sono in grado di attivare vie diverse con effetti diversi (3). Il signaling mediato dall’ERα promuove la proliferazione cellulare mentre quello di ERβ la inibisce (probabilmente per un effetto anche pro-apoptotico)(4).
Studi successivi hanno dimostrato un ruolo del complesso E2/ERα nella trascrizione di geni chiave per la proliferazione, differenziazione, sopravvivenza delle cellule sane, ma anche per l’invasività, la metastatizzazione e l’angiogenesi delle cellule neoplastiche (c-myc, ciclina D, A ed E, p21)(5), mentre ERβ inibisce la trascrizione ERα-mediata e la proliferazione E2-mediata nelle cellule di cancro mammario bloccando le cellule allo stadio G2 (6).
Circa il 70% dei cancri della mammella esprime gli ER, ma, a differenza delle cellule mammarie sane, esprimono più ERα che ERβ; inoltre possono esprimere il recettore del progesterone (PR) e l’erythroblastosis oncogene-B2 (ErbB-2, HER2/neu). Per tutti questi motivi, già da molti anni sono in uso e in studio nella terapia del carcinoma della mammella numerosi farmaci che agiscono sul sistema estrogenico.
Agonisti del GnRH
Nelle donne in pre- e peri-menopausa può esserne indicato l’utilizzo per sopprimere la produzione estrogenica ovarica. Questi farmaci si sono dimostrati efficaci al pari dell’annessiectomia bilaterale sulla sopravvivenza totale (OS) e libera da insuccesso (FFS)(7).
I farmaci che presentano come indicazione il carcinoma della mammella sono il triptorelin ed il leuprorelin. Nelle donne di età superiore ai 40 anni che vengono sottoposte a chemioterapia anti-neoplastica standard si verifica amenorrea permanente in quasi il 100% dei casi, per cui l’utilizzo degli analoghi del GnRH in queste pazienti va discusso, mentre l’associazione di chemioterapia tradizionale e castrazione farmacologica sembra essere l’approccio ottimale nelle donne in pre-menopausa più giovani (8,9).
Selective estrogen-receptor modulators (SERM)
Sono una classe di ligandi dell’ER che agiscono come anti-estrogeni o agonisti recettoriali, in base al tessuto che esprime il recettore; a livello mammario agiscono come anti-estrogeni.
Il capostipite è il tamoxifene, già in commercio da circa 30 anni, che viene generalmente somministrato per 5 anni dalla diagnosi e si è dimostrato in grado di ridurre il rischio di recidiva (RR 0.53 a 5 anni, 0.68 a 10 anni e 0.97 a 15 anni)(10), determinando un aumento di circa il 15% della sopravvivenza libera da recidiva (RFS)(11). Probabilmente la riduzione dell’effetto a lungo termine è associata a un meccanismo di resistenza, che può essere associato a isoforme dell’ER che non legano il farmaco o all’aumentata attività di co-attivatori oppure ad alterazioni di tappe della via di trasmissione del segnale intra-cellulare (12). È indicato in tutte le pazienti con carcinoma mammario ER+ e/o PR+. Il tamoxifene per agire deve essere metabolizzato dal citocromo P450-2D6: circa il 6-8% delle donne sono portatrici di forme meno attive di questo enzima e farmaci come gli SSRI lo inibiscono.
Altri membri di questa famiglia sono il raloxifene e il più nuovo toremifene, indicato nel carcinoma mammario metastatico nel post-menopausa.
Tra i possibili rischi di questi farmaci ci sono l’iperplasia endometriale fino al carcinoma dell’endometrio, per l’effetto agonista sull’utero, ed eventi trombo-embolici.
Inibitori dell'aromatasi (AI)
L’aromatasi è un’enzima che catalizza la reazione di trasformazione degli androgeni in estrogeni (estrone ed E2). È normalmente espresso in molti tessuti, compresa la mammella. Nella donna in post-menopausa la principale fonte di E2 non è più l’ovaio, ma sono i tessuti che contengono l’aromatasi, in particolare il tessuto adiposo.
Gli AI hanno come indicazione la terapia endocrina del carcinoma della mammella per 5 anni nelle donne in post-menopausa.
Membri di questa famiglia sono anastrozolo, letrozolo, exemestane. Vengono metabolizzati dal citocromo P450-3A4, per cui bisogna porre attenzione alle interazioni con farmaci induttori e inibitori di questo enzima (13).
Tra gli effetti indesiderati più importanti c’è la progressione dell’osteoporosi, per cui le pazienti devono effettuare una densitometria ossea prima e durante il trattamento e spesso è necessario associare una terapia con bisfosfonati.
Lo studio ATAC (Arimidex, Tamoxifen, Alone or in Combination) ha posto a confronto l’anastrozolo con il tamoxifene in donne in post-menopausa con carcinoma della mammella ER+: dopo un follow-up di 68 mesi, lo studio ha evidenziato che l’anastrozolo prolungava la sopravvivenza libera da malattia (DFS), il tempo alla recidiva, e riduceva le metastasi a distanza, con minori effetti secondari del tamoxifene, in particolare a livello ginecologico e cardiovascolare, suggerendo quindi di preferire gli AI ai SERM nelle donne in post-menopausa (14). Risultati simili sono stati ottenuti anche da uno studio successivo di confronto tra letrozolo e tamoxifene (15).
Selective estrogen-receptor down-regulators (SERD)
Sono una classe di steroidi puramente anti-estrogeni. L’unico farmaco di questa classe attualmente in uso è il fulvestrant, indicato nel trattamento in post-menopausa nelle donne in ricaduta di malattia durante o dopo terapia anti-estrogenica adiuvante. Presenta come potenziale rischio il tromboembolismo venoso. Il farmaco è in commercio in Italia, ma sottoposto a monitoraggio da parte dell’AIFA (13,16). Uno studio di confronto con il tamoxifene non ha evidenziato, nei pazienti ER+, differenze significative per i principali esiti (14,17).
Ligandi di ERβ
Non esistono ancora farmaci in commercio che agiscano da agonisti del recettore ERβ, ma, in considerazione dei suoi effetti anti-proliferativi e pro-apoptotici, potrebbe essere una classe di farmaci promettente per il trattamento del carcinoma della mammella.
Nelle pazienti che sovra-esprimono HER2 (che spesso sono anche resistenti alla terapia ormonale) è possibile associare all’eventuale terapia endocrina adiuvante il trastuzumab o il lapatinib, anticorpi monoclonali che legano il recettore HER2/neu, che normalmente lega il fattore di crescita dell’epidermide (EGF), determinando l’attivazione di diverse vie di segnale che promuovono la proliferazione cellulare e l’angiogenesi (18). Il lapatinib agisce inibendo l’attività tirosin-chinasica legata al recettore dell’EGF (EGF-R) e a HER2/neu.
Diversi studi hanno anche valutato l’utilizzo di questi approcci come terapia neo-adiuvante nel carcinoma della mammella: uno studio ha evidenziato la capacità del letrozolo di determinare un 55% di risposta tumorale oggettiva vs il 35% del tamoxifene e in circa il 50% dei casi è stato possibile evitare la mastectomia radicale, optando per una quadrantectomia (19).
L’IGF-1 e il suo recettore influenzano lo sviluppo e la crescita tumorale. L’IGF-1R appare sovra-espresso nella maggioranza dei carcinomi della mammella (90-95%) ed è spesso co-espresso con ER. Gli estrogeni inducono l’espressione di IGF-1R. L’obesità e dunque l’iperinsulinemia riducono la produzione di IGFBP-1 e IGFBP-2 (IGF-binding proteins) che normalmente legano e inibiscono l’azione dell’IGF-1; questo potrebbe essere uno dei motivi della maggior prevalenza di carcinoma mammario nelle donne obese (13,20). Per questo motivo sono state sviluppate molecole inibenti IGF-1R, come linsitinib, e sono al momento in fase di sperimentazione clinica per diversi tipi di neoplasie (21).
È inoltre interessante come diversi studi abbiano attribuito un effetto anti-neoplastico alla metformina: uno studio inglese ha evidenziato che l’uso continuo per almeno 5 anni della metformina ha determinato un odds ratio di 0.44 per lo sviluppo di carcinoma della mammella rispetto alle donne non in trattamento con metformina, probabilmente per un effetto indiretto di riduzione dei livelli circolanti di insulina (22).
Nuovi target terapeutici
Tra i nuovi target terapeutici, di particolare interesse sono i co-attivatori e co-repressori delle vie di trasduzione del segnale estrogenico: tra i co-attivatori, SRC-1 e SRC-3 sono frequentemente sovra-espressi nel carcinoma della mammella, soprattutto in quelli con peggiore prognosi. Il gossipolo, un prodotto naturale estratto dal seme del cotone, sembra essere in grado di down-regolare l’espressione di SRC-3 nelle cellule di carcinoma mammario; antagonizza mediatori anti-apoptotici e quindi rappresenta un possibile prototipo di molecole agenti sul sistema estrogenico (21,23).
L’espressione del recettore ERα può essere modulata in seguito all’esposizione a un inibitore dell’enzima istone-deacetilasi (HDAC) (Vorinostat, Belinostat, Panobinostat). Queste molecole sono in grado di bloccare il ciclo cellulare e di indurre apoptosi in molte cellule neoplastiche. Inoltre, questi farmaci sembrerebbero anche permettere la ricomparsa di ERα nelle cellule neoplastiche precedentemente ER- e determinano inoltre una riduzione dell’espressione dell’EGF-R, risensibilizzando le cellule alla terapia endocrina (24,25).
Sono in fase di studio per nuovi farmaci numerosi altri target molecolari delle complesse vie di trasduzione del segnale di ERα (PI3K/AKT, mTOR, Src, HSP90, …) (13).
Il cancro della prostata rappresenta la neoplasia più comune nell’uomo ed è dipendente dal sistema androgenico. Questa correlazione è nota a partire dagli anni ’70, da quando venne notata una regressione del carcinoma metastatico in uomini sottoposti a castrazione medica o chirurgica (26). Tuttavia, si notò subito come la deprivazione androgenica non era curativa, in quanto in molti uomini la neoplasia avanzava e portava a decesso nonostante livelli molto bassi di testosterone, con una sopravvivenza media di 2-3 anni (27).
I meccanismi di resistenza alla deprivazione androgenica possono essere molteplici: aumentata espressione di enzimi coinvolti nella steroidogenesi, aumentata espressione del recettore per gli androgeni (AR), mutazioni genetiche di AR o diversa specificità di ligando, attivazione di vie di segnale mediate da AR senza legame con il ligando, attivazione di vie indipendenti che possono superare le vie indotte dal blocco androgenico, proliferazione di cellule staminali prostatiche indipendenti dagli androgeni (28). È proprio la comprensione di questi meccanismi che ha portato allo sviluppo di farmaci sempre più nuovi e sempre più specifici per contrastare lo sviluppo di neoplasie cosiddette resistenti alla castrazione (CRPC). Inoltre, è interessante notare come in alcuni pazienti che sviluppano resistenza, in particolare nei pazienti in terapia con flutamide, la sospensione del farmaco determina una riduzione paradossa dei valori di PSA: questo potrebbe essere attribuito a una mutazione dell’AR, che farebbe comportare il farmaco come un agonista piuttosto che come antagonista. Pertanto, potrebbe essere utile in alcuni pazienti che presentano un rialzo del PSA in terapia ormonale adiuvante, tentare la sospensione del farmaco e ricontrollare a breve il PSA (29).
La terapia endocrina del carcinoma della prostata è indicata (29):
- come terapia adiuvante nei pazienti a rischio basso (T1-T2a, Gleason ≤ 6, PSA 10 anni in associazione o meno alla radioterapia;
- nei pazienti a rischio intermedio (T2b-T2c, G > 6 o PSA 10-20 ng/mL) con metastasi linfonodali in associazione o meno alla radioterapia;
- nei pazienti ad alto rischio (T3a o G 8-10 o PSA > 20 ng/mL), a rischio molto alto (T3b-T4) e metastatici (M1), in associazione alla radioterapia.
Agonisti e antagonisti del GnRH
Sono utilizzati in alternativa all’orchiectomia bilaterale per ottenere un’efficace deprivazione di androgeni determinando una castrazione farmacologica.
I farmaci che hanno indicazione per il carcinoma della prostata sono goserelin, triptorelin e leuprorelin e il più nuovo degarelix, che blocca invece l’interazione del GnRH con il suo recettore a livello ipofisario, inducendo una più rapida castrazione ed evitando l’iniziale aumento del testosterone associato all’inizio della terapia con gli analoghi (tuttavia necessita di somministrazioni mensili ed è associato a frequenti reazioni nel sito di inoculazione) (30). La castrazione ottenuta con questi farmaci è equivalente all’orchiectomia e non sembrerebbero esserci grandi differenze tra i vari farmaci (31). Inoltre, questi farmaci in associazione alla radioterapia aumentano significativamente la DFS rispetto alla sola radioterapia (32).
Tra gli effetti secondari più importanti sono la ginecomastia e i sintomi da ipogonadismo.
Ketoconazolo
Blocca la sintesi di androgeni, ma anche di corticosteroidi e mineralcorticoidi, per cui andrebbe somministrato in associazione a terapia corticosteroidea sostitutiva. Al momento non è utilizzato e la sua utilità è controversa.
Abiraterone acetato
È un potente inibitore della sintesi degli androgeni, agendo come potente e selettivo inibitore dell’enzima CYP17 (17α-idrossilasi/C17,20-liasi). Uno studio di fase III in uomini con CRPC avanzato, trattati in precedenza con chemioterapia tradizionale, ha evidenziato un aumento della sopravvivenza mediana rispetto al placebo. Il farmaco è commercializzato da aprile 2013 in Italia (con il nome di Zytiga)(33). È il primo farmaco endocrino a dimostrare un’efficacia nei CRPC (si parla infatti di terapia ormonale secondaria). Anche uno studio su pazienti CPRC naïve da chemioterapia tradizionale ha dimostrato una migliore PFS radiologica e un trend di aumento dell’OS rispetto al placebo (34).
Il farmaco determina un aumento di ACTH, che potrebbe determinare un eccesso di mineralcorticoidi, dal momento che quella è l’unica via della steroidogenesi non bloccata; questo potrebbe determinare un quadro di iperaldosteronismo primitivo, controllabile con l’aggiunta di prednisone 5 mg/die (30).
Orteronel (TAK-700)
È un inibitore selettivo di CYP17, con maggiore selettività per l’attività 17,20-liasica piuttosto che per la 17-idrossilasi. Evita la co-somministrazione del corticosteroide importante nella terapia con abiraterone (anche se a dosaggi elevati l’orteronel può a sua volta sopprimere la sintesi di corticosteroidi). I primi dati di fase I e II hanno evidenziato una buona efficacia del farmaco, sia in termini di riduzione dei livelli di testosterone e di DHEAS, sia di riduzione del PSA.
Galeterone (TOK-001)
Oltre a essere un inibitore di CYP17, blocca competitivamente il legame con gli androgeni e down-regola in vitro l’espressione dell’AR. Il farmaco è ancora in fase sperimentale.
Anti-androgeni non steroidei
Sono in uso da molti anni, i principali sono bicalutamide e flutamide. Si legano al dominio legante il ligando dell’AR (LBD). Sono efficaci nell’indurre il blocco androgenico, in associazione agli agonisti del GnRH. La loro somministrazione andrebbe iniziata almeno 3 giorni prima dell’inizio degli agonisti del GnRH, per attenuarne l’effetto di rialzo transitorio del testosterone.
Tra gli effetti secondari più importanti, la dolenzia mammaria e la ginecomastia, il calo della libido e la disfunzione erettile.
Diversi studi hanno valutato l’efficacia di questi farmaci in monoterapia o associati alla castrazione farmacologica: uno studio ha confrontato bicalutamide 150 mg vs la castrazione farmacologica o chirurgica ed ha evidenziato un’equivalenza dei due approcci nei pazienti con tumore localmente avanzato, ma non nei metastatici, in cui l’associazione determina risultati migliori (35). Una metanalisi ha riportato un maggior rischio di recidiva nei pazienti trattati con anti-androgeni in monoterapia piuttosto che in associazione con la castrazione farmacologica (36). Quindi le linee guida suggeriscono di effettuare il blocco androgenico combinato, mentre la monoterapia con gli anti-androgeni può essere discussa per la minore tossicità e per il minor effetto “ipogonadizzante” rispetto al blocco combinato con gli analoghi del GnRH.
Bloccanti le vie di trasduzione dell’AR
L’enzalutamide (MDV3100) è un anti-androgeno non steroideo con altissima affinità per AR, che, oltre ad inibire il recettore stesso, ne blocca la traslocazione nel nucleo. Negli studi di fase I e II nei pazienti con CRPC ha determinato una riduzione del PSA > 50% e una risposta radiologica nel 22% dei pazienti; gli studi di fase III vs placebo hanno evidenziato una maggiore sopravvivenza mediana nei pazienti trattati, con una riduzione del 37% del rischio decesso (37-39).
ARN-509 è un analogo dell’enzalutamide ma più potente: inibisce specificamente la crescita delle cellule che esprimono l’AR; impedisce anch’esso la traslocazione nucleare del recettore. È ancora in fase I e II di sperimentazione.
EPI-001
A differenza della maggior parte degli anti-androgeni che si legano al LBD, questo nuovo farmaco si lega al dominio N-terminale (NTD) e al dominio di legame del DNA (DBD), inibendo il legame del recettore attivato o transattivato al DNA, impedendo la trascrizione genica indipendentemente dal legame del ligando al recettore, by-passando quindi alcuni dei principali meccanismi di sviluppo di CRPC (30).
Il carcinoma dell’endometrio si suddivide in due tipi:
- endometrioide tipo 1 (90% dei casi, a basso grado di malignità) associato a fattori di rischio ormonali ben riconosciuti;
- endometrioide tipo 2 (ad alto grado di malignità).
Tutte le condizioni che causano un’eccessiva esposizione agli estrogeni endogeni o esogeni determinano uno stimolo proliferativo sull’endometrio, che a lungo termine può indurre iperplasia e quindi neoplasia endometriale. Il progesterone, invece, a differenza dell’effetto proliferativo sulla mammella, ha un effetto anti-mitogeno sull’endometrio e riduce la proliferazione cellulare.
Tutte le condizioni che causano iperestrogenismo in associazione ad anovularietà causano una stimolazione endometriale, in assenza di una controparte progestinica. Un esempio è l’obesità. Tumori ovarici e surrenalici che producono estrogeni rappresentano condizioni di rischio per lo sviluppo di carcinoma dell’endometrio. Altri fattori sono la nulliparità, la menopausa tardiva, la familiarità di primo grado, la terapia ormonale sostitutiva post-menopausa e la terapia con SERM nel carcinoma della mammella (40).
Una revisione Cochrane ha analizzato quale potesse essere il ruolo della terapia ormonale sostitutiva post-menopausa nell’insorgenza dell’iperplasia dell’endometrio: lo studio ha evidenziato un rischio aumentato nelle pazienti in terapia con soli estrogeni, mentre il rischio si azzerava nelle pazienti trattate anche con progestinici (41). Per l’uso di soli estrogeni il rischio di iperplasia e di carcinoma aumenta con il tempo: 1.30 a 2 anni, 4.50 da 5-10 anni. Per quanto riguarda l’uso di progestinici in schemi sequenziali, l’ideale è non somministrarli per meno di 10 giorni al mese, perché questo incrementa il rischio di cancro dell’endometrio (42). Per quanto riguarda il rischio di carcinoma endometriale nelle pazienti in terapia con SERM, alcuni studi non hanno evidenziato un rischio significativo con l’uso del raloxifene a differenza del tamoxifene (43).
Nell’ambito della terapia del carcinoma dell’endometrio, la terapia endocrina trova un posto limitato rispetto agli altri tumori ormono-sensibili già discussi: i progestinici sono in grado di ridurre l’espressione degli ER, riducono la trascrizione di geni coinvolti nella crescita cellulare ER-dipendente, attivano il gene onco-soppressore p21. Attualmente, sono disponibili progestinici di quarta generazione (dienogest), che non presentano attività androgenica, riducendo quindi tutti gli effetti secondari associati all’attività androgenica dei progestinici di prima, seconda e terza generazione. Inoltre, questi sembrano avere proprietà anti-angiogenetiche (44).
Anche per il carcinoma dell’endometrio, come per la mammella, sembrano promettenti farmaci di nuova generazione, come gli inibitori di mTOR (everolimus), gli inibitori dell’istone-deacetilasi (HDACi) e un ruolo interessante è svolto sempre dalla metformina, che sembra indurre l’espressione di PR sulle cellule endometriali, attraverso l’inibizione di IGF-1 e 2; promettente potrebbe quindi essere il suo uso in associazione a un progestinico.
BIBLIOGRAFIA
- Southam CM. The complex etiology of cancer. Cancer Res 1963, 23: 1105-15.
- Moore DH, et al. Breast carcinoma etiological factors. Adv Cancer Res 1983, 40: 189-253.
- Saville B, et al. Ligand-, cell-, and estrogen receptor subtype (alpha/beta)-dependent activation at GC-rich (Sp1) promoter elements. J Biol Chem 2000, 275: 5379–87.
- Helguero LA. Estrogen receptors alfa (ERa) and beta (ERb) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 2005, 24: 6605–16.
- Musgrove EA, et al. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011, 11: 558–72.
- Ström A, et al. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci USA 2004, 101: 1566–71.
- Taylor CW, et al. Multicenter randomized clinical trial of goserelin versus surgical ovariectomy in premenopausal patients with receptor-positive metastatic breast cancer: an intergroup study. J Clin Oncol 1998, 16: 994-9.
- Goodwin PJ, et al. Risk of menopause during the first year after breast cancer diagnosis. J Clin Oncol 1999, 17: 2365-70.
- Goel S, et al. LHRH agonists for adjuvant therapy of early breast cancer in premenopausal women. Cochrane Database Syst Rev 2009: CD004562.
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011, 378: 771-84.
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG). Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 1998, 351: 1451-67.
- Clarke R, et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22: 7316–39.
- Renoir JM, et al. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol 2013, 85: 449–465.
- Howell A, et al. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years' adjuvant treatment for breast cancer. Lancet 2005, 365: 60-2.
- Coates AS, et al. Five years of letrozole compared with tamoxifen as initial adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer: update of study BIG 1-98. J Clin Oncol 2007, 25: 486-92.
- www.agenziafarmaco.gov.it
- Howell A, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol 2004, 22: 1605-13.
- Ménard S, et al. Biologic and therapeutic role of HER2 in cancer. Oncogene 2003, 22: 6570–8.
- Eiermann W, et al. Preoperative treatment of postmenopausal breast cancer patients with letrozole: A randomized double-blind multicenter study. Ann Oncol 2001, 12: 1527-32.
- Patterson RE, et al. Metabolism and breast cancer risk: frontiers in research and practice. J Acad Nutr Diet 2013, 113: 288-96.
- www.clinicaltrials.gov
- Bodmer M, et al. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care 2010, 33: 1304-8.
- Wang Y, et al. Small molecule inhibition of the steroid receptor coactivators: SRC-3 and SRC-1. Mol Endocrinol 2011, 25: 2041–53.
- Urbinati G, et al. Liposomes loaded with histone deacetylase inhibitors for breast cancer therapy. Int J Pharm 2010, 397: 184–93.
- Zhou Q, et al. Histone deacetylase inhibitor LBH589 reactivates silenced estrogen receptor alpha (ER) gene expression without loss of DNA hypermethylation. Cancer Biol Ther 2007, 6: 64–9.
- Huggins C, Hodges CV. Studies on prostatic cancer. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin 1972, 22: 232–40.
- Halabi S, et al. Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol 2003, 21: 1232–7.
- Amaral TM, et al. Castration-resistant prostate cancer: mechanisms, targets, and treatment. Prostate Cancer 2012, 2012: 327253.
- NCCN Clinical Practice Guidelines for Prostate Cancer 2013, www.nccn.org.
- Friedlander TW, Ryan CJ. Targeting the androgen receptor. Urol Clin North Am 2012, 39: 453-64.
- Seidenfeld J, et al. Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. Ann Intern Med 2000, 132: 566-77.
- Pilepich MV, et al. Androgen suppression adjuvant to definitive radiotherapy in prostate carcinoma--long-term results of phase III RTOG 85-31. Int J Radiat Oncol Biol Phys 2005, 61: 1285-90.
- De Bono JS, et al. Circulating tumor cells predict survival benefit from treatment in metastatic castration-resistant prostate cancer. Clin Cancer Res 2008, 14: 6302–9.
- Ryan CJ. Interim analysis (IA) results of COU-AA-302, a randomized, Phase III study of abiraterone acetate (AA) in chemotherapy-naive patients (pts) with metastatic castration-resistant prostate cancer (mCRPC), American Society of Clinical Oncology Annual Meeting. Chicago, June 2, 2012.
- Iversen P, et al. Bicalutamide monotherapy compared with castration in patients with nonmetastatic locally advanced prostate cancer: 6.3 years of follow up. J Urol 2000, 164: 1579-82.
- Samson DJ, et al. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer 2002, 95: 361-76.
- Scher HI, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet 2010, 375: 1437-46.
- Scher HI, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012, 367: 1187-97.
- Tsao CK, et al. Targeting the androgen receptor signalling axis in castration-resistant prostate cancer (CRPC). BJU Int 2012, 110: 1580-8.
- Amant F, et al. Endometrial cancer. Lancet 2005, 366: 491-505.
- Furness S, et al. Hormone therapy in postmenopausal women and risk of endometrial hyperplasia. Cochrane Database Syst Rev 2012: CD000402.
- Pike MC, et al. Estrogen-progestin replacement therapy and endometrial cancer. J Natl Cancer Inst 1997, 89: 1110-6.
- Martino S, et al. Continuing outcomes relevant to Evista: breast cancer incidence in postmenopausal osteoporotic women in a randomized trial of raloxifene. J Natl Cancer Inst 2004, 96: 1751-61.
- Umene K, et al. New candidate therapeutic agents for endometrial cancer: potential for clinical practice (review). Oncol Rep 2013, 29: 855-60.
Il carcinoma della mammella ormono-sensibile
Gabriele Luppi & Elena Barbieri
Oncologia Medica, Azienda Ospedaliero-Universitaria di Modena
La neoplasia della mammella è quella con maggior frequenza e la seconda causa di morte per tumore nel sesso femminile [1].
Circa il 70% dei tumori della mammella esprime positività per il recettore per gli estrogeni e/o per il progesterone. Il recettore per gli estrogeni è il principale fattore di trascrizione che guida l’oncogenesi nelle neoplasie mammarie a recettori ormonali positive ed HER2 negative ed inoltre è un fattore predittivo di risposta alle terapie con anti-estrogeni.
Storicamente, il trattamento ormonale del carcinoma della mammella rappresenta uno dei primi esempi di terapia personalizzata in oncologia da quando, più di un secolo fa, Sir George Beatson osservò tre casi di carcinoma della mammella in fase avanzata che regredirono dopo ovariectomia [2], aprendo la strada all’utilizzo della manipolazione ormonale nel carcinoma della mammella.
Ad oggi la quantità di conoscenze acquisite ha permesso di identificare sottogruppi che differiscono dal punto di vista prognostico e predittivo di risposta alle terapie, rendendo il carcinoma della mammella l’esempio più tipico della sfida di personalizzare la terapia sulla base di caratteristiche peculiari della patologia e delle pazienti.
Perou et al. [3] individuarono profili molecolari che caratterizzano i carcinomi della mammella e ne permettono la classificazione in base a profili di espressione genica. In base a questa classificazione, i carcinomi mammari che esprimono i recettori ormonali si possono suddividere ulteriormente in due gruppi, Luminal A e Luminal B, che differiscono tra loro dal punto di vista genico e molecolare, con ricadute sul piano prognostico e predittivo [4]. Oltre a questi gruppi, ne sono stati identificati ulteriori due che tipicamente non esprimono i recettori ormonali e che corrispondono ai sottogruppi basal-like e HER2-enriched.
La tecnica dei micro-arrays, e di conseguenza la suddivisione in sottogruppi in base ai profili di espressione genica, non è ancora entrata nella pratica clinica di tutti i giorni e pertanto si è cercato di identificare tramite immunoistochimica caratteristiche proprie dei vari sottogruppi molecolari.
I tumori Luminal A sono caratterizzati dalla forte espressione dei recettori ormonali e da un basso indice di proliferazione (Ki-67 < 14%). Sono generalmente ad ottima prognosi, traggono scarso beneficio dall’aggiunta della chemioterapia alla manipolazione ormonale, che rimane il caposaldo del trattamento di queste neoplasie, e tipicamente hanno un andamento indolente, presentando recidive tardive (spesso ben oltre i 5-10 anni dalla diagnosi) e principalmente a livello osseo.
I tumori Luminal B presentano espressione meno marcata dei recettori ormonali e sovente mancano di espressione del recettore per il progesterone, hanno un’elevata velocità di proliferazione o la concomitante espressione della proteina HER2. Dal punto di vista clinico e sulla base delle sole caratteristiche biologiche, sono guidati nella crescita anche da altri fattori e non solo dalla via estrogenica, hanno un comportamento clinico più aggressivo, beneficiano della chemioterapia e tendono a recidivare più precocemente e soprattutto a livello viscerale [5].
I farmaci ad azione ormonale utilizzati nel trattamento del carcinoma della mammella possono essere classificati in base al meccanismo d’azione. I modulatori selettivi del recettore per gli estrogeni (SERMs), come il tamoxifene ed il raloxifene, sono agonisti/antagonisti del recettore per gli estrogeni. A livello del tessuto ghiandolare mammario, il tamoxifene agisce come antagonista, col risultato di interrompere la trascrizione dei geni la cui espressione è regolata dagli estrogeni e quindi il loro effetto di stimolo alla proliferazione. Il fulvestrant agisce anch’esso a livello del recettore per gli estrogeni, ma, a differenza del tamoxifene, è dotato solamente di attività antagonista, poiché porta alla degradazione del recettore estrogenico causandone la perdita.
Per indurre una deprivazione estrogenica le strategie in uso includono:
- nelle donne in pre-menopausa la soppressione della funzionalità ovarica (chirurgica, radioterapica o farmacologica);
- nelle donne in post-menopausa l’utilizzo di inibitori dell’aromatasi.
La scelta del tipo di terapia ormonale si basa su una serie di fattori, che includono lo stato menopausale ed il profilo di tossicità atteso.
Negli ultimi tempi sta emergendo l’utilizzo della terapia ormonale anche in fase pre-operatoria per le pazienti con malattia a recettori ormonali positivi/HER2 negativa. Questo tipo di approccio, finora riservato quasi esclusivamente alle pazienti anziane, sta assumendo sempre maggiore interesse nel campo della ricerca e parte dall’osservazione che in fase adiuvante, alle pazienti con neoplasia a recettori ormonali positivi ed HER2 negativa viene offerto generalmente il solo trattamento ormonale.
L’opportunità offerta dalla terapia sistemica primaria di valutare i biomarcatori prima e dopo il trattamento permette di identificare precocemente le pazienti che più probabilmente trarranno beneficio dal trattamento ormonale. Ad esempio è stato generato un punteggio prognostico (preoperative prognostic index, PEPI score) che si basa sulla dimensione tumorale, sul coinvolgimento linfonodale, sull’espressione del Ki-67 e del recettore per gli estrogeni secondo Allred dopo la terapia ormonale sistemica primaria [6].
Nonostante quanto detto in precedenza, alcune pazienti mostrano comunque una resistenza primaria o acquisita alla terapia ormonale, suggerendo di fatto il ruolo del cross-talk tra i recettori ormonali e altre vie molecolari intra-cellulari che portano a resistenza nei confronti della terapia endocrina. Bloccando entrambe le vie coinvolte, combinando la terapia ormonale con terapie a bersaglio molecolare, si può potenziare l’attività anti-tumorale e ristabilire la sensibilità all’ormono-terapia.
Ad esempio, in uno studio randomizzato di terapia pre-operatoria in donne con malattia a recettori ormonali positive e con espressione di EGFR, la monoterapia con l’inibitore tirosin-chinasico gefitinib ha comportato una riduzione delle dimensioni tumorali nel 54% delle pazienti, mentre la combinazione gefitinib + anastrozolo vs. gefitinib da solo ha permesso di inibire maggiormente la proliferazione tumorale [7].
Esperienze simili sono state condotte sia in fase metastatica, sia in fase neoadiuvante, aggiungendo alla terapia ormonale con letrozolo, lapatinib, un duplice inibitore di EGFR/HER2 ad attività tirosin-chinasica [8,9].
E’ stato dimostrato che la via di segnalazione intra-cellulare legata a PI3K/Akt/mTOR è legata alla resistenza alla terapia endocrina [10]. L’inibitore di mTOR everolimus ha aumentato in maniera significativa l’efficacia di letrozolo nel setting neoadiuvante: il tasso di risposta clinica nel braccio di combinazione letrozolo/everolimus è risultato maggiore rispetto al solo letrozolo (68.1% vs. 59.1%). Inoltre, dopo 15 giorni di trattamento si è osservata una riduzione dell’espressione di Ki-67 nel 57% di pazienti trattate con l’aggiunta di everolimus rispetto al 30% del braccio con placebo [11].
Nell’ambito della malattia metastatica recenti studi hanno dimostrato il beneficio dell’aggiunta di everolimus al trattamento ormonale in donne in post-menopausa progredite o recidivate dopo il trattamento con un’inibitore dell’aromatasi non steroideo (anastrozolo o letrozolo). Lo studio BOLERO-2 ha riportato un incremento di 2.4 volte della progression-free survival mediana con la combinazione di everolimus + exemestane vs. exemestane da solo [12]. Lo studio TAMRAD ha dimostrato un miglioramento in termini di beneficio clinico, tempo a progressione e sopravvivenza globale nelle pazienti trattate con everolimus + tamoxifene vs. tamoxifene da solo [13].
A più di un secolo dalla scoperta del suo ruolo nel trattamento del carcinoma mammario, la terapia endocrina ottimale per il carcinoma della mammella è in continua evoluzione. Le ricerche volte a caratterizzare in maniera sempre più precisa i carcinomi mammari ormono-sensibili perseguono l’obiettivo di personalizzare le terapie in base alle caratteristiche della malattia fino a livello molecolare, ed a quelle individuali di ogni paziente.
Bibliografia
- Siegel R, Naishadham D, Jemal A. Cancer Statistics 2012. CA Cancer J Clin 2012, 62: 10-29.
- Beatson GW. On the treatment of inoperable cases of carcinoma of the mamma: suggestion of a new method of treatment with illustrative cases. The Lancet 1896, 2: 162-5.
- Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumor. Nature 2000, 406: 747-52.
- Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst 2009, 101: 736-50.
- Kennecke H, Yerushalmi R, Woods R, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol 2010, 28: 3271-7.
- Ellis MJ, Tao Y, Luo J, et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J Natl Cancer Inst 2008, 100: 1380-8.
- Polychronis A, Sinnett HD, Hadjiminas D, et al. Preoperative gefitinib versus gefitinib and anastrozole in postmenopausal patients with oestrogen-receptor positive and epidermal-growth-factor-receptor-positive primary breast cancer: a double-blind placebo-controlled phase II randomised trial. Lancet Oncol 2005, 383-91.
- Johnston S, Pippen J Jr, Pivot X, et al. Lapatinib combined with Letrozole and placebo as first-line therapy for postmenopausal hormone receptor positive metastatic breast cancer. J Clin Oncol 2009, 27: 5538-46.
- Conte PF, Guarneri V, Generali DG, et al. Double-blind, placebo-controlled, multicentric randomized phase IIb neoadjuvant study of letrozole-lapatinib in postmenopausal HER2-negative, hormone receptor-positive operable breast cancer. J Clin Oncol 2011, 29 suppl: abstr 550.
- Tokunaga E, Kimura Y, Mashino K, et al. Activation of PI3K/Akt signaling and hormone resistance in breast cancer. Breast Cancer 2006, 13: 137-44.
- Baselga J, Semiglazov V, van Dam P, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol 2009, 27: 2630-7.
- Baselga J, Campone M, Piccart M, et al. Everolimus for postmenopausal hormone receptor-positive advanced breast cancer. N Engl J Med 2012, 366: 520-9.
- Bachelot T, Bourgier C, Cropet C, et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol 2012, 30: 2718-24.
Carcinoma endometriale
Enrico Vizza, Benito Chiofalo, Valentina Bruno
UOC Ginecologia Oncologica, Istituto Nazionale Tumori "Regina Elena-IFO"
(aggiornato al 18 dicembre 2020)
EPIDEMIOLOGIA E FATTORI DI RISCHIO
Il carcinoma dell’endometrio è una neoplasia tipica dell’età post-menopausale: in oltre il 90% dei casi la diagnosi avviene dopo i 50 anni, con età media di 63 anni.
Nei Paesi economicamente più sviluppati rappresenta la neoplasia ginecologica più frequente, in Italia è il 5° tumore maligno più frequente nelle donne.
I principali fattori di rischio possono essere suddivisi in tre categorie.
Fattori ambientali: l’obesità e un regime alimentare ricco di grassi animali sono elementi comuni nell’anamnesi patologica delle pazienti affette da carcinoma dell’endometrio, essendo responsabili di un’aumentata produzione di estrogeni a seguito dell’aromatizzazione degli androgeni di provenienza surrenalica (1). In menopausa, quando la produzione estrogenica ovarica è praticamente nulla, il deidroepiandrosterone surrenalico viene convertito dagli adipociti in estrone, che ha un’azione stimolante sulle cellule endometriali. Altri fattori di rischio sono l’ipertensione arteriosa, il diabete mellito e le epatopatie croniche. Al contrario, attività fisica e diete ricche di fibre sembrano fattori protettivi (2).
Fattori ormonali: la presenza di un’attività estrogenica, sia endogena che esogena, non contro-bilanciata da una corretta quantità di progesterone sembra una delle principali cause di neoplasia endometriale. L’allungamento della finestra estrogenica (menarca precoce, menopausa tardiva e nulliparità) aumenta il rischio di sviluppare questa neoplasia. Una condizione clinica predisponente sembra la policistosi ovarica, soprattutto in un quadro di sindrome metabolica, a causa di cicli anovulatori, iperestrogenismo relativo e iperandrogenismo. Anche i tumori estrogeno-secernenti, come i tumori ovarici dei cordoni sessuali (cellule della granulosa, ecc), rappresentano fattori di rischio. Molti studi hanno messo in evidenza come il rischio di tumore dell’endometrio aumenti da 2 a 10 volte a seguito di terapia ormonale sostitutiva estrogenica in climaterio, senza l’associazione di progestinici (3-4). Le attuali linee guida per il trattamento dei disturbi associati alla menopausa raccomandano, quindi, la terapia estrogenica associata a progestinici nelle donne non sottoposte a pregressa isterectomia, mentre l’estrogeno può essere impiegato da solo nelle donne precedentemente isterectomizzate. Il rischio associato al trattamento con tamoxifene, agonista parziale degli estrogeni, è dibattuto in letteratura: secondo alcuni autori il pregresso carcinoma mammario è di per sé un fattore di rischio per la neoplasia endometriale, indipendentemente dal tipo di terapia ormonale utilizzata (5). Numerose evidenze scientifiche hanno infine mostrato che l’utilizzo di combinazioni estro-progestiniche in età fertile rappresenta un fattore protettivo, determinando una riduzione del rischio di tumore dell’endometrio di circa il 50%, con un effetto protettivo che si prolunga per più di 20 anni dopo la sospensione.
Fattori ereditari: esistono infine forme ereditarie di carcinoma endometriale. Nella sindrome di Lynch di tipo II, autosomica dominante, la predisposizione allo sviluppo di neoplasie è dovuta all’alterazione di geni che appartengono al sistema del mismatch repair (MMR). Oltre a una maggiore predisposizione genetica al solo carcinoma endometriale, è possibile una predisposizione familiare a sviluppare tumori maligni in diversi altri organi (6): il rischio è del 40-80% per tumore del colon, 40-60% per carcinoma dell’endometrio e 10-12% per carcinoma ovarico. È stato proposto che l’insorgenza in una donna giovane di una neoplasia endometriale possa essere considerato come evento sentinella della sindrome di Lynch.
ISTOPATOLOGIA
I tumori endometriali derivano dalle cellule ghiandolari di derivazione mulleriana. Nel 1983, Bokhman formulò l’ipotesi dell’esistenza di due varianti di carcinoma endometriale con diversa patogenesi:
- tipo I: comprende l’adenocarcinoma endometrioide, che rappresenta l’80% dei casi, è estrogeno-dipendente, insorge frequentemente su iperplasia endometriale e ha prognosi generalmente favorevole;
- tipo II: comprende l’adenocarcinoma sieroso-papillifero (< 10% dei casi) e l’adenocarcinoma a cellule chiare (2-4% dei casi). Non sono estrogeno-dipendenti, insorgono solitamente su endometrio atrofico, colpiscono soggetti più anziani e hanno elevata aggressività biologica, con prognosi sfavorevole (7).
Nel 2013 la caratterizzazione genomica di 373 campioni di carcinoma endometriale ha consentito una nuova classificazione biomolecolare più complessa, successivamente sviluppata nel tempo, con importanti risvolti diagnostici e terapeutici (8).
Dal punto di vista anatomo-patologico il carcinoma endometriale si presenta con differenti istotipi.
- Adenocarcinoma endometrioide: è l’istotipo più frequente (80%) ed è estrogeno-correlato. È generalmente puro, tuttavia può essere associato alla presenza di un carcinoma non endometrioide e la proporzione delle componenti influenza la diffusione della malattia e la prognosi.
- Adenocarcinoma sieroso-papillare: rappresenta circa il 5-10% dei carcinomi dell’endometrio e va sospettato nelle donne in una fascia di età di 10 anni superiore a quella dell’adenocarcinoma endometrioide. Spesso di alto grado, presenta solitamente un’infiltrazione miometriale profonda e particolare tropismo per i linfonodi. Diffonde rapidamente alla cavità peritoneale ed è pertanto a prognosi infausta (9).
- Adenocarcinoma a cellule chiare: è molto più raro dell’adenocarcinoma sieroso, rappresentando circa l’1% dei carcinomi dell’endometrio, è tipico dell’età avanzata e ha prognosi sfavorevole. Il carcinoma a cellule chiare tipico, per definizione, ha gli stessi caratteri istologici architetturali del carcinoma a cellule chiare di altre sedi genitali (10).
- Adenocarcinoma mucinoso: rappresenta circa l’1% dei tumori endometriali e va distinto dall’adenocarcinoma endo-cervicale primitivo, da cui si differenzia per l’abbondante presenza di mucina.
- Carcinoma squamoso: rappresenta meno dell’1% dei tumori endometriali e va distinto dall’adenocarcinoma a differenziazione squamosa (per l’assenza di differenziazione ghiandolare) e dai carcinomi a cellule squamose della cervice uterina diffusi a livello endometriale.
- Tumori misti: rappresentano meno dell’1% dei tumori endometriali e le diverse componenti superano ciascuna il 10% del totale.
- Carcinoma indifferenziato: sono tumori composti da masse solide di cellule indifferenziate. Possono essere associati ad adenocarcinoma endometrioide o rappresentare l’unica componente documentata nel tumore. Più rari sono i carcinomi indifferenziati a piccole cellule, simili a quelli di altri organi, che mostrano differenziazione neuroendocrina con positività per cromogranina, sinaptofisina e altri tipici marcatori.
- Carcinosarcoma: si compone di elementi sia sarcomatosi che carcinomatosi.
- Sarcoma endometriale stromale: quelli di alto grado sono molto aggressivi, con una sopravvivenza a 5 anni che va da 0 a 55%.
Oltre all’istotipo, i principali fattori di rischio per la comparsa di metastasi e quindi di un esito sfavorevole sono: il grado di differenziazione, il pattern molecolare, il grado di interessamento miometriale, l’invasione degli spazi linfatici e vascolari, l’interessamento cervicale e la diffusione extra-uterina della neoplasia.
Le sedi di più frequente localizzazione secondaria sono polmone, fegato, osso (prevalentemente vertebre) ed encefalo.
CLINICA E DIAGNOSI
L’esordio clinico del carcinoma endometriale è rappresentato, nella grande maggioranza dei casi, da un sanguinamento vaginale anomalo: perdita ematica vaginale in menopausa o sanguinamento inatteso, rispetto al flusso mestruale normale, in età fertile. Raramente invece la neoplasia decorre in maniera asintomatica e la diagnosi viene posta in modo accidentale.
In caso di sanguinamenti uterini anomali, il primo esame da eseguire è l’ecografia trans-vaginale per lo studio dell’utero e degli annessi, al fine di valutare lo spessore endometriale (sospetto se > 4 mm in menopausa) ed eventuali neoformazioni intra-uterine (11). L’aggiunta del Doppler (color e power) rende possibile studiare con accuratezza la perfusione ematica dell’endometrio e dei processi espansivi a suo carico (12). L’ecografia trans-vaginale eseguita da operatori esperti riesce a valutare con alta sensibilità l’infiltrazione miometriale, cervicale ed extra-uterina.
La diagnosi di carcinoma endometriale si basa essenzialmente sulla valutazione istologica del tessuto endometriale ottenuto attraverso prelievi bioptici.
Il gold standard per lo studio dell’endometrio è l’isteroscopia diagnostica, che consente la visualizzazione del canale cervicale e della cavità uterina, permettendo allo stesso tempo l’esecuzione di biopsie mirate (13-14).
Per una stadiazione clinica, la risonanza magnetica (RM) pelvica con mezzo di contrasto è in grado di valutare accuratamente il grado di infiltrazione miometriale (sensibilità 87%), l’infiltrazione dello stroma cervicale (sensibilità 80%) e delle pareti vaginali. Permette inoltre di valutare l’infiltrazione del tessuto parametriale e la presenza di linfadenomegalie pelviche o lombo-aortiche, con accuratezza del 76% (15).
La tomografia computerizzata (TC) presenta scarsa accuratezza diagnostica rispetto alla RM nella valutazione dell’infiltrazione miometriale, ma risulta utile nella valutazione delle eventuali sedi extra-uterine di malattia.
La tomografia ad emissione di positroni (PET) con 18F-fluorodeossiglucosio può essere impiegata sia per la rilevazione di metastasi linfonodali (N) durante la valutazione pre-chirurgica che nel follow-up post-operatorio (16).
La scintigrafia ossea è limitata invece ai casi clinicamente sospetti per localizzazione ossea.
Ulteriori indagini (cistoscopia, colonscopia, ecc) sono eseguite su indicazione clinica.
La stadiazione del carcinoma endometriale è anatomo-chirurgica. Quella attualmente utilizzata è la stadiazione FIGO 2009 (tabella), che è fondamentale per la decisione dell’iter terapeutico.
| Stadiazione del carcinoma endometriale | ||
| Stadio | Caratteristiche | |
| I | Tumore limitato al corpo del’utero | |
| Ia | Nessuna infiltrazione o < metà del miometrio | |
| Ib | Infiltrazione > metà del miometrio | |
| II | Tumore esteso allo stroma cervicale ma non fuori dell’utero | |
| III | Estensione locale o regionale | |
| IIIa | Alla sierosa uterina o alle ovaie | |
| IIIb | Alla vagina o alla regione parametriale | |
| IIIc | A linfonodi pelvici (IIIc1) o lombo-aortici (IIIc2, indipendentemente dai linfonodi pelvici) | |
| IV | Estensione alla mucosa vescicale o intestinale o metastasi a distanza | |
| IVa | Alla mucosa vescicale o intestinale | |
| IVb | Metastasi a distanza | |
TRATTAMENTO
Trattamento chirurgico
L’approccio chirurgico standard, nei primi stadi di malattia, è rappresentato dall’isterectomia totale extra-fasciale e dall’annessiectomia bilaterale, con o senza linfadenectomia pelvica e/o lombo-aortica.
Le possibili modalità di accesso chirurgico sono la via laparotomica, laparoscopica, robotica e vaginale. Numerosi studi hanno confrontato l’approccio laparotomico con quello laparoscopico, dimostrando che, a parità di sopravvivenza globale a 5 anni in entrambi i gruppi, la laparoscopia era associata a numerosi benefici in termini di durata dell’ospedalizzazione, complicanze, controllo del dolore e qualità di vita (17). Per quanto riguarda l’approccio mini-invasivo robotico, è riportato un numero di complicanze significativamente minore rispetto alla via laparotomica; inoltre, nelle pazienti obese, la chirurgia robotica sembra superiore anche all’approccio laparoscopico, associandosi a minore perdita ematica, minor tasso di conversione laparotomica, ridotti tempi operatori e durata della degenza (18-19).
Il ruolo della linfadenectomia in questo gruppo di pazienti è tuttora oggetto di discussione. Da uno studio italiano randomizzato (20), condotto su 514 pazienti con carcinoma dell’endometrio in stadio I, è emerso che la linfadenectomia non era associata a tassi maggiori di sopravvivenza libera da malattia (PFS). Un’ulteriore conferma di questi dati è derivata dallo studio ASTEC, in cui la linfadenectomia pelvica non ha dimostrato un beneficio in termini di sopravvivenza globale (OS) e intervallo di tempo libero da malattia (21). Il limite di questi studi è tuttavia rappresentato dall’inclusione di una popolazione in cui il rischio di presentare metastasi linfonodali era troppo basso per evidenziare un effetto positivo della linfadenectomia sulla sopravvivenza. Rimane pertanto irrisolto il ruolo della linfadenectomia nelle pazienti ad alto rischio. Alcuni autori suggeriscono di considerare una stadiazione chirurgica completa nelle pazienti con rischio intermedio-alto (stadio IA G3 e IB sec. FIGO 2009) per identificare quelle pazienti che richiedono un trattamento adiuvante post-operatorio. Lo studio SEPAL, pubblicato nel 2010, ha invece valutato l’estensione della linfadenectomia, confrontando la linfadenectomia pelvica vs linfadenectomia pelvica e lombo-aortica (22). Dai dati emersi, le pazienti ad alto rischio hanno beneficiato di una linfadenectomia pelvica e lombo-aortica in termini di OS. Altri studi randomizzati sono in corso per valutare il ruolo della linfadenectomia, con lo scopo di chiarire quale sia l’iter terapeutico più efficace per le pazienti ad alto rischio.
La biopsia del linfonodo sentinella ha mostrato una buona performance diagnostica e rappresenta un buon compromesso tra l’esecuzione di una stadiazione chirurgica completa e l’omissione di una linfadenectomia sistematica. In particolare, la biopsia del linfonodo sentinella è in grado di ridurre la morbilità potenzialmente derivabile dall’esecuzione di una linfadenectomia sistematica, mentre il suo “ultrastaging”, ovvero la sezione microscopica in più fette per una diagnosi più accurata, consente l’individuazione di micro-metastasi linfonodali spesso non diagnosticate con l’esame istologico convenzionale (anche in pazienti considerate a basso rischio). Uno studio osservazionale multicentrico sul linfonodo sentinella ha riportato una sensibilità dell’84% e un valore predittivo negativo del 97% in donne con carcinoma endometriale in stadio I-II sec. FIGO (23). La maggior parte degli studi pubblicati su questo argomento ha previsto l’identificazione del linfonodo sentinella mediante iniezione cervicale di un tracciante come il verde indocianina.
Trattamento conservativo
Nelle giovani donne desiderose di prole, affette da carcinoma endometriale con caratteristiche prognostiche favorevoli, come istotipo endometrioide, ben differenziato, invasione miometriale minima/assente, può essere ipotizzato un trattamento conservativo che preveda la somministrazione di un progestinico orale (medrossiprogesterone o megestrolo acetato) o endo-uterino (spirale levonorgestrel-medicata), associato o meno alla somministrazione di analoghi del GnRH. Il trattamento farmacologico può essere preceduto o meno da chirurgia resettoscopica. Il trattamento conservativo è da considerarsi tuttavia temporaneo e finalizzato all’ottenimento della gravidanza, che deve essere fortemente incoraggiata a partire dal riscontro di una risposta tumorale completa. Il trattamento chirurgico standard è comunque raccomandato in caso di progressione di malattia, mancata risposta tumorale completa a 12 mesi o al termine della gravidanza.
Trattamenti complementari e adiuvanti
Nella malattia diffusa (stadio III e IV), alcuni autori suggeriscono di considerare il massimo sforzo chirurgico nelle pazienti con un buon performance status, data la scarsa risposta ai regimi chemioterapici. Qualora la chirurgia non sia fattibile a causa di contro-indicazioni mediche (5-10% delle pazienti), oltre al trattamento medico con carboplatino e paclitaxel, può essere preso in considerazione un trattamento radioterapico, con o senza brachiterapia, per il controllo locale di malattia.
Dopo la chirurgia, in base alla presenza dei diversi fattori prognostici (età, stadio della malattia, istologia, grading, profondità di infiltrazione miometriale, interessamento degli spazi vascolari, coinvolgimento linfonodale), le pazienti vengono suddivise in classi di rischio per decidere se effettuare eventuali trattamenti adiuvanti (24).
La radioterapia ha un ruolo importante nel trattamento del carcinoma endometriale. Può essere utilizzata come trattamento adiuvante (radioterapia esterna con o senza brachiterapia) nelle pazienti ad alto rischio dopo la chirurgia, come terapia esclusiva nelle pazienti in cui la procedura chirurgica è improponibile, nelle pazienti con recidiva di malattia in associazione alla chemioterapia o come terapia palliativa negli stadi avanzati (25).
Anche per quanto riguarda il trattamento chemioterapico esistono diversi regimi attuabili a seconda dell’istotipo, stadio e classi di rischio.
Terapia ormonale
Il carcinoma endometriale può esprimere sia recettori per estrogeni (ER) che per progesterone (PR) e quindi può rispondere a una terapia sistemica ormonale.
La terapia ormonale è raccomandata solo per l’istologia endometrioide, in quanto neoplasia ormono-sensibile, e include, primariamente, l’uso di agenti progestinici. Vengono anche utilizzati il tamoxifene e gli inibitori dell'aromatasi sebbene vi siano meno evidenze (26-27).
Nel trattamento adiuvante non è stato dimostrato, all’interno di sette studi randomizzati, alcun vantaggio in termini di riduzioni del rischio di morte per malattia nelle donne trattate con ormonoterapia con progestinico verso la sola osservazione. Tuttavia, nelle pazienti sottoposte a trattamento ormonale vi è stata un’incidenza del 5% di eventi trombo-embolici (28).
Il trattamento ormonale viene quindi indicato nel trattamento della recidiva di malattia/malattia metastatica dei carcinomi ad istologia endometrioide, in particolare nelle pazienti non candidabili a chemioterapia sistemica. I fattori predittivi di risposta al trattamento nella malattia metastatica includono: tumori ben differenziati, lunga PFS, localizzazione metastatiche extra-pelviche (soprattutto polmonari) ed espressione di PR e/o ER (26,27,29-31). Quest’ultimo dato ha portato, nella pratica clinica, analogamente alle recidive di carcinoma mammario, a considerare una nuova procedura bioptica per determinare l’assetto recettoriale ormonale nelle pazienti con recidive di malattia, soprattutto se con lungo intervallo di PFS (32).
Più recentemente l’ormonoterapia nell’endometrio ha trovato un ulteriore spazio di utilizzo nel trattamento conservativo, in pazienti giovani e desiderose di prole, del carcinoma endometrioide stadio IA G1. Una recente revisione ha riportato un tasso di risposte patologiche complete durature variabile tra 57 e 76% con progesterone somministrato per via orale o uso topico con la spirale medicata, a fronte di una percentuale di non responders e di pazienti che vanno in progressione durante trattamento del 12-25% e di una quota di pazienti variabile tra il 25 e il 30% che dopo una iniziale risposta sviluppa una recidiva di malattia a un tempo mediano di 19 mesi (27,33).
I progestinici sono impiegati nel trattamento del carcinoma endometriale dagli anni ’50, con tassi di risposta obiettiva variabili tra l’11-56% e una PFS compresa tra i 2.5 e i 14 mesi (34). Una revisione più recente ha mostrato tassi di risposta variabili dal 15 al 30% e sopravvivenze globali di 7-11 mesi nel trattamento con progestinici nelle pazienti con recidiva di malattia o malattia metastatica (35).
Gli effetti collaterali legati al trattamento sono di solito minori: edemi, aumento ponderale. Da considerare l’aumentato rischio di TVP nelle pazienti con anamnesi positiva per tale patologia o con patologie della coagulazione.
Alte dosi di megestrolo o medrossiprogesterone acetato (MAP) non aumentano l’efficacia terapeutica, come dimostrato in uno studio randomizzato (36), che ha comparato MAP 200 mg/die verso 1000 mg/die. Rimane quindi raccomandata la dose di MAP 200 mg/die o megestrolo 160 mg/die (26).
Nei diversi studi clinici le risposte sono per la maggior parte parziali e di breve durata, nonostante alcune pazienti abbiano risposte di lunga durata (36-37).
Allo scopo di contrastare la resistenza ai trattamenti ormonali, è stata testata la combinazione di temsirolimus (inibitore di mTOR) con o senza megestrolo e tamoxifene nel trattamento dei carcinomi dell’endometrio avanzati o recidivati. Lo studio non ha dimostrato alcun vantaggio della combinazione ormonoterapia e temsirolimus, andando invece a incrementare gli eventi avversi, tra cui la TVP (38). L’attività di temsirolimus è, invece, preservata nelle pazienti sottoposte a precedente chemioterapia sistemica.
Altre terapie ormonali hanno dimostrato attività nei carcinomi endometriali in stadio avanzato o recidivato.
Per quanto concerne gli inibitori dell'aromatasi, i dati derivati da studi retrospettivi e studi di fase II si sono dimostrati molto modesti e contrastanti (39,40). Questi farmaci possono essere considerati in prima o seconda linea nelle pazienti non candidabili a trattamento chirurgico o con recidiva di malattia e che non possono ricevere progestinici (41). L’efficacia del trattamento non sembra correlata con l’espressione dei recettori ormonali o con mutazioni di determinate vie di segnale molecolare (42). Un recente studio di fase II (43) con la combinazione di letrozolo ed everolimus in pazienti pretrattate ha mostrato un alto tasso di beneficio clinico (40%) e di risposta (32%). In uno studio di fase II della Nordic Society of Gynecologic Oncology l’exemestane ha mostrato un tasso di risposta del 10%, mancata progressione di malattia dopo 6 mesi nel 35% delle pazienti trattate, con buon profilo di tossicità globale (44).
BIBLIOGRAFIA
- Crosbie EJ, Zwahlen M, Kitchener HC, et al. Body mass index, hormone replacement therapy, and endometrial cancer risk: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2010, 19: 3119-30.
- World Cancer Research Fund/American Institute for Cancer Research. Diet, nutrition, physical activity, and prevention of cancer: a global perspective. Continuous Update Project Exper Report 2018.
- International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risks to hormones: vol. 72 Hormonal contraception and post-menopausal hormonal therapy. IARC Lyon, 1999.
- Bruno V, Corrado G, Baci D, et al. Endometrial cancer immune escape mechanisms: let us learn from the fetal-maternal interface. Front Oncol 2020, 10: 156.
- Chiofalo B, Mazzon I, Di Angelo Antonio S, et al. Hysteroscopic evaluation of endometrial changes in breast cancer women with or without hormone therapies: results from a large multicenter cohort study. J Minim Invasive Gynecol 2020, 27: 832‐9.
- Garg K, Shih K, Barakat R, et al. Endometrial carcinomas in women aged 40 years and younger: tumors associated with loss of DNA mismatch repair proteins comprise a distinct clinicopathologic subset. Am J Surg Pathol 2009, 33: 1869-77.
- Bockman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol 1983, 15: 10-7.
- Kommoss S, McConechy MK, Kommoss F, et al. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann Oncol 2018, 29: 1180–8.
- Alkushi A, Kobel M, Kalloger SE, et al. High-grade endometrial carcinoma: serous and grade 3 endometrioid carcinomas have different immunophenotypes and outcomes. Int J Gynecol Pathol 2010, 29: 343-50.
- Cirisano FD, Robboy SJ, Dodge RK, Gilks CBl. The outcome of stage I-II clinically and surgically staged papillary serous and clear cell endometrial cancers when compared with endometrioid carcinoma. Gynecol Oncol 2000, 77: 55-65.
- Savelli L, Ceccarini M, Ludovisi M, et al. Preoperative local staging of endometrial cancer: transvaginal sonography vs. magnetic resonance imaging. Ultrasound Obstet Gynecol 2008, 31: 560-6.
- Epstein E, Van Holsbeke C, Mascilini F, et al. Gray-scale and color Doppler ultrasound characteristics of endometrial cancer in relation to stage, grade and tumor size. Ultrasound Obstet Gynecol 2011, 38: 586-93.
- Vitale SG, Bruni S, Chiofalo B, et al. Updates in office hysteroscopy: a practical decalogue to perform a correct procedure. Updates Surg 2020, 72: 967-76.
- Chiofalo B, Palmara V, Vilos GA, et al. Reproductive outcomes of infertile women undergoing "see and treat" office hysteroscopy: a retrospective observational study. Minim Invasive Ther Allied Technol 2019, Dec 19: 1‐7.
- Manfredi R, Mirk P, Maresca G, et al. Loco-regional staging of endometrial carcinoma: role of MR imaging in predicting surgical staging. Radiology 2004, 231: 372-8.
- Suga T, Nakamoto Y, Saga T, et al. Clinical value of FDG-PET for preoperative evaluation of endometrial cancer. Ann Nucl Med 2011, 25: 269-75.
- Walker JL, Piedmonte MR, Spirtos NM, et al. Recurrence and survival after random assignment to laparoscopy versus laparotomy for comprehensive surgical staging of uterine cancer: Gynecologic Oncology Group LAP2 Study. J Clin Oncol 2012, 30: 695-700.
- Burke WM, Gossner G, Goldman NA, et al. Robotic surgery in the obese gynecologic patient. Clin Obstet Gynecol 2011, 54: 420-30.
- Vizza E, Chiofalo B, Cutillo G, et al. Robotic single site radical hysterectomy plus pelvic lymphadenectomy in gynecological cancers. J Gynecol Oncol 2018, 29: e2.
- Benedetti Panici P, Basile S, Maneschi F, et al. Systematic pelvic lymphadenectomy vs no lymphadenectomy in early-stage endometrial carcinoma: randomized clinical trial. J Natl Cancer Inst 2008, 100: 1707-16.
- ASTEC study group, Kitchener H, Swart AM, Qian Q, et al. Efficacy of systematic pelvic lymphadenectomy in endometrial cancer (MRC ASTEC trial): a randomised study. Lancet 2009, 373: 125-36.
- Todo Y, Kato H, Kaneuchi M, et al. Survival effect of para-aortic lymphadenectomy in endometrial cancer (SEPAL study): a retrospective cohort analysis. Lancet 2010, 375: 1165-72.
- Ballester M, Dubernard G, Lécuru F, et al. Detection rate and diagnostic accuracy of sentinel-node biopsy in early stage endometrial cancer: a prospective multicentre study (SENTI-ENDO). Lancet Oncol 2011, 12: 469-76.
- Vizza E, Cutillo G, Bruno V, et al. Pattern of recurrence in patients with endometrial cancer: a retrospective study. Eur J Surg Oncol 2020, 46: 1697-702.
- Creutzberg CL, Nout RA, Lybeert MLM, et al. Fifteen-year radiotherapy outcomes of the randomized PORTEC-1 trial for endometrial carcinoma. Int J Radiat Oncol Biol Phys 2011, 81: 631–8.
- Colombo N, et al. Endometrial cancer: ESMO clinical practice guidelines. Ann Oncol 2013, 24 suppl 6: vi33-8.
- Linee guida AIOM. Neoplasie dell’utero, endometrio e cervice. Edizione 2019.
- Martin-Hirsch PP, Bryant A, Keep SL, et al. Adjuvant progestagens for endometrial cancer. Cochrane Database Syst Rev 2011, 6: CD001040.
- Singh M, et al. Relationship of estrogen and progesterone receptors to clinical outcome in metastatic endometrial carcinoma: a Gynecologic Oncology Group Study. Gynecol Oncol 2007, 106: 325-33.
- Chambers JT, et al. Estrogen and progestin receptor levels as prognosticators for survival in endometrial cancer. Gynecol Oncol 1988, 31: 65-81.
- Piver MS, et al. Medroxyprogesterone acetate (Depo-Provera) vs. hydroxyprogesterone caproate (Delalutin) in women with metastatic endometrial adenocarcinoma. Cancer 1980, 45: 268-72.
- Tangen IL, et al. Loss of progesterone receptor links to high proliferation and increases from primary to metastatic endometrial cancer lesions. Eur J Cancer 2014, 50: 3003–10.
- Bovicelli A, D'Andrilli G, Giordano A, De Iaco P. Conservative treatment of early endometrial cancer. J Cell Physiol 2013, 228: 1154-8.
- Decruze SB, Green JA. Hormone therapy in advanced and recurrent endometrial cancer: a systematic review. Int J Gynecol Cancer 2007, 17: 964-78.
- Kokka F, Brockbank E, Oram D, et al. Hormonal therapy in advanced or recurrent endometrial cancer. Cochrane Database Syst Rev 2010, 12: CD007926.
- Thingpen JT, et al. Oral medroxyprogesterone acetate in the treatment of advanced or recurrent endometrial carcinoma: a dose-response study by the Gynecologic Oncology Group. J Clin Oncol 1999, 17: 1736-44.
- Crespo C, et al. Metastatic endometrial cancer in lung and liver: complete and prolonged response to hormonal therapy with progestins. Gynecol Oncol 1999, 72: 250-5.
- Fleming GF, et al. Temsirolimus with or without megestrol acetate and tamoxifen for endometrial cancer: a Gynecologic Oncology Group Study. Gynecol Oncol 2014, 132: 585-92.
- Steed HL, Chu QS. Aromatase inhibition: a potential target for the management of recurrent or metastatic endometrial cancer by letrozole: more questions than answers? Expert Opin Investig Drugs 2011, 20: 681-90.
- Bogliolo S, Gardella B. Effectiveness of aromatase inhibitors in the treatment of advanced endometrial adenocarcinoma. Arch Gynecol Obstet 2016, 293: 701-8.
- Altman AD, Thompson J, Nelson G, et al. Use of aromatase inhibitors as first- and second-line medical therapy in patients with endometrial adenocarcinoma: a retrospective study. J Obstet Gynaecol Can 2012, 34: 664-72.
- Ma BB, Oza A, Eisenhauer E, et al. The activity of letrozole in patients with advanced or recurrent endometrial cancer and correlation with biological markers — a study of the National Cancer Institute of Canada Clinical Trials Group. Int J Gynecol Cancer 2004, 14: 650–8.
- Slomovitz BM, et al. Phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J Clin Oncol 2015, 33: 930-6.
- Lindemann K, Malander S, Christensen RD, et al. Examestane in advanced or recurrent endometrial carcinoma: a prospective phase II study by the Nordic Society of Gynecologic Oncology (NSGO). BMC Cancer 2014, 14: 68.
Sindromi paraneoplastiche endocrine
Ipercalcemia paraneoplastica
Fabio Vescini
SOC Endocrinologia e Malattie del Metabolismo, Azienda Ospedaliero-Universitaria S. Maria della Misericordia, Udine
L’ipercalcemia si può presentare in circa il 10% dei pazienti con carcinoma in stadio avanzato e, spesso, rappresenta un fattore prognostico sfavorevole, gravato da una mortalità di circa il 50% a 30 giorni dall’insorgenza. Questa forma di ipercalcemia è generalmente caratterizzata da una ridotta concentrazione di paratormone (PTH), al contrario di quella che si osserva nell’iperparatiroidismo primitivo, perché è conservato ed efficace il feed-back del calcio plasmatico sulle paratiroidi (1-2).
I meccanismi principali attraverso i quali un tumore può provocare ipercalcemia sono essenzialmente quattro (1-4).
- Produzione di sostanze in grado di mimare l’azione del PTH. Questa forma, che rappresenta circa l’80% delle ipercalcemie paraneoplastiche, è definita ipercalcemia neoplastica umorale (HHM) ed è generalmente sostenuta dalla produzione, da parte del tumore primitivo, o, più frequentemente, dalle metastasi tumorali, del peptide correlato al PTH (PTHrP). Le neoplasie in grado di produrre PTHrP sono quelle del polmone (squamocellulare), del rene, della mammella, del capo e collo (sempre forme a cellule squamose), della vescica, dell’utero e dell’ovaio. Occasionalmente anche i tumori neuroendocrini possono secernere PTHrP e, ancora più raramente, i sarcomi e alcune neoplasie ematologiche.
- Metastasi ossee osteolitiche, in grado di mobilizzare direttamente il calcio mediante un marcato incremento del riassorbimento osseo osteclasto-mediato. Esse rappresentano circa il 20% delle ipercalcemie paraneoplastiche e, in molti casi, sono sostenute da neoplasie della mammella, mieloma multiplo e linfomi.
- Produzione tumorale di vitamina D, ovvero sovra-espressione di 1-alfa-idrossilasi nel tessuto neoplastico, con conseguente aumento della produzione di calcitriolo; tale condizione è stata descritta in associazione a certi linfomi.
- Produzione metastatica di citochine con attività osteoclastogenica, ovvero del PTH vero e proprio (casi molto rari).
La HHM è sicuramente la forma più comune. Il PTHrP è un peptide monomerico, che esiste in diverse isoforme, di lunghezza variabile da 60 a 173 aminoacidi (5-6). Molti tessuti presentano una fisiologica, ancorché minima, produzione di PTHrP, ma il suo ruolo fisiologico non è stato ancora del tutto compreso. Dati sperimentali mostrano che il PTHrP regola l’ossificazione encondrale, controlla la proliferazione delle cartilagini articolari, promuove il differenziamento osteoblastico nel microambiente osseo e sicuramente partecipa al controllo del passaggio placentare del calcio (la delezione del gene è letale per i feti dei mammiferi)(5-6). In corso di HHM il PTHrP incrementa notevolmente e può agire in maniera sovrapponibile al PTH, legandosi ai recettori renali e ossei, dove è in grado di incrementare la calcemia e ridurre il riassorbimento tubulare di fosfati (1).
I pazienti affetti da HHM presentano la sintomatologia tipica dell’ipercalcemia, caratterizzata da nausea, vomito, letargia, insufficienza renale e, se non adeguatamente trattata, coma e morte. La gravità dei sintomi dipende sia dalla concentrazione plasmatica del calcio, sia dalla rapidità d’insorgenza dell’ipercalcemia, con le forme acute che possono rappresentare una vera e propria emergenza medica (1,2).
Il laboratorio mostra tutte le alterazioni tipiche dell’iperparatiroidismo primitivo (oltre all’ipercalcemia, ipofosforemia, iperfosfaturia, ipercalciuria ed elevazione della fosfatasi alcalina), ma con valori di PTH soppressi o inappropriatamente normali e, comunque, generalmente < 20-30 pg/mL. Anche in pazienti con PTH superiore a questa soglia, ma all’interno del range di normalità, in presenza di ipercalcemia deve essere sempre sospettata la presenza di una HHM, in modo particolare se si tratta di soggetti anziani, con fattori di rischio neoplastico o anamnesi positiva per pregresse neoplasie (1-3). In alcuni laboratori è possibile dosare il PTHrP, generalmente con metodiche immunometriche, ma va tenuto presente che, viste l’elevata concentrazione plasmatica di questo peptide nella HHM, è possibile avere dei falsi negativi a causa del cosiddetto “effetto gancio” (risultati artificialmente bassi ad alte concentrazioni di analita)(7).
Il trattamento ottimale dell’ipercalcemia paraneoplastica è, ovviamente, rappresentato dalla rimozione della neoplasia causale; tuttavia, non è infrequente l’impossibilità di procedere a questo tipo di approccio chirurgico e, in questo caso, devono essere adottate terapie mediche adeguate (tabella).
| Farmaci per il trattamento dell’ipercalcemia paraneoplastica | ||
| Via di somministrazione | Posologia | |
| Soluzione fisiologica | ev | 200-500 mL/ora |
| Furosemide | ev | 20-40 mg/die |
| Pamidronato | ev | 60-90 mg (ripetibile dopo 3-4 settimane) |
| Zoledronato | ev | 4 mg (ripetibile dopo 3-4 settimane) |
| Prednisone | po (nel mieloma e nei linfomi) | 40-100 mg/die |
| Calcitonina | sc/im | 4-8 UI/kg/12 ore |
Innanzi tutto è sempre raccomandabile rivalutare la terapia generale seguita dal paziente e, se possibile, sospendere i farmaci potenzialmente ipercalcemizzanti (supplementi di calcio, litio, diuretici tiazidici, anti-acidi a base di calcio, vitamina D)(1).
L’approccio di prima linea all’ipercalcemia neoplastica è rappresentato dalla terapia re-idratante con soluzione salina, al fine di aumentare la filtrazione glomerulare e, contemporaneamente, inibire il riassorbimento tubulare del calcio. Solo quando il volume plasmatico è stato ristabilito, potranno essere aggiunti alla terapia i diuretici dell’ansa, tenendo sempre sotto controllo la concentrazione plasmatica del potassio (1).
Il trattamento medico di scelta, tuttavia, è quello con bisfosfonati, sia per la loro efficacia nel ridurre la calcemia, sia per il loro profilo di sicurezza. La somministrazione endovenosa di pamidronato o zoledronato induce una diminuzione significativa della calcemia in 2-4 giorni, raggiungendo un nadir intorno ai 7 giorni dopo l’infusione. L’effetto ipocalcemizzante è sufficientemente duraturo, potendo persistere anche per 3-4 settimane (1,4).
Nel caso in cui non si possano utilizzare i bisfosfonati (es. insufficienza renale, pre-esistente osteonecrosi della mandibola), si potrà ricorrere alla calcitonina, ancorché il suo effetto ipocalcemizzante è minore e di durata inferiore rispetto ai bisfosfonati (8).
Solo pochi casi selezionati, trattati in centri ad elevatissima specializzazione, potranno giovarsi di una terapia con mitramicina ovvero con nitrato di gallio (8).
Infine nei pazienti con insufficienza renale e cardiaca, che non possono essere trattati con elevate quantità di liquidi e con bisfosfonati, andrà preso in considerazione il trattamento emodialitico (4).
Bibliografia
- Pelosof LC, Gerber DE. Paraneoplastic syndromes: an approach to diagnosis and treatment. Mayo Clin Proc 2010, 85: 838-54.
- Guise TA, Mundy GR. Cancer and bone. Endocr Rev 1998, 19: 18-54.
- Jacobs TP, Bilezikian JP. Rare causes of hypercalcemia. J Clin Endocrinol Metab 2005, 90: 6316-22.
- Stewart AF. Hypercalcemia associated with cancer. N Engl J Med 2005, 352: 373-9.
- Mundy GR, Edwards JR. PTH-related peptide (PTHrP) in hypercalcemia. J Am Soc Nephrol 2008, 19: 672-5.
- Clemens TL, Cormier S, Eichinger A, et al. Parathyroid hormone-related protein and its receptors: nuclear functions and roles in the renal and cardiovascular systems, the placental trophoblasts and the pancreatic islets. Br J Pharmacol 2001, 134: 1113-36.
- Burtis WJ. Parathyroid hormone-related protein: structure, function, and measurement. Clin Chem 1992, 38: 2171-83.
- Lumachi F, Brunello A, Roma A, et al. Medical treatment of malignancy-associated hypercalcemia. Curr Med Chem 2008, 15: 415-21.
Ipoglicemia paraneoplastica
Ettore Seregni
SS Terapia Medico Nucleare ed Endocrinologia, Fondazione IRCCS Istituto Nazionale Tumori, Milano
In ambito oncologico il riscontro di ipoglicemia rappresenta un evento relativamente frequente. Una riduzione delle concentrazioni ematiche di glucosio può essere osservata, ad esempio, nei pazienti con neoplasie maligne che coinvolgono in maniera estesa e diffusa il parenchima epatico o surrenalico. In queste condizioni inadeguata gluconeogenesi e ridotta secrezione di ormoni glucocorticoidi sostengono il quadro metabolico. Inoltre, l’ipoglicemia è di frequente riscontro negli stadi terminali del malato oncologico e, ancora, severe crisi ipoglicemiche caratterizzano i tumori secernenti insulina (insulinomi pancreatici e tumori ectopici secernenti insulina).
Tuttavia, l’ipoglicemia paraneoplastica non è in senso stretto riconducibile alle cause sovramenzionate e, in particolare, alla presenza di un tumore insulare. La presenza di ipoglicemia in assenza di tumore insulare viene definita con l’acronimo di NICTH (da Non-Islet Cell Tumor Hypoglycemia)(1). Per definizione, quindi la NICTH non è sostenuta dalla produzione di insulina.
La NICHT è una condizione clinicamente molto rara (incidenza di circa 1 caso per milione/anno) e si stima che l’ipoglicemia da NICHT sia almeno quattro volte meno frequente di quella attribuibile alla presenza di un tumore insulare (insulinoma).
La sindrome di Douge-Potter (descritta in maniera indipendente dai due Autori nel 1930) rappresenta uno dei primi riscontri di ipoglicemia paraneoplastica e deriva dall’associazione di NICTH con un particolare tipo di neoplasia, il tumore fibroso solitario (SFT, acronimo di Solitary Fibrous Tumor)(2,3). Questo tipo di tumore mesenchimale insorge a livello intra-toracico dalla pleura polmonare e presenta un comportamento benigno nella maggior parte dei casi. In circa il 20% dei casi, invece, il tumore mostra un comportamento maligno, con recidive locali e metastasi a distanza, per lo più a livello epatico. Inoltre, sono descritti SFT a insorgenza extra-toracica, più frequentemente in sede retro-peritoneale peri-vescicale. Istologicamente, il tumore si caratterizza per la presenza di cellule fibroblastiche o miofibroblastiche fusiformi e per una tipica architettura vascolare di tipo emangiopericitoma, con positività per CD34. È da sottolineare, però, che il SFT si associa a NICTH solo nel 5-10% dei casi (4-7).
Oltre alla sindrome di Douge-Potter, la NICTH può riscontrarsi anche in altre neoplasie maligne: epatocarcinomi, tumori germinali, tumori stromali gastro-intestinali (GIST) e anche neoplasie di origine ematopoietica (8-14).
L’ipoglicemia riconosce come meccanismo fisio-patologico la produzione e il rilascio da parte delle cellule tumorali di un precursore a elevato peso molecolare dell’IGF-II (big IGF-II)(1,15,16). In condizioni normali la big IGF-II corrisponde a circa al 10-20% dell’IGF-II totale, mentre nelle situazioni di NICTH questa quota supera generalmente il 60%. L’eccesso di big IGF-II conduce a ipoglicemia attraverso esagerata stimolazione dei recettori bersaglio, quali i recettori per IGF-1 (IGF1-R) e per insulina (IR), che comporta aumentata captazione di glucosio da parte delle cellule muscolari e adipose, e riduzione della gluconeogenesi epatica. L’iperstimolazione di IGF1-R e IR è sostenuta dal fatto che la big IGF-II, a differenza dell’IGF-II, non forma complessi ternari sufficientemente stabili con le proteine leganti specifiche (IGFBP) e con la subunità acido-labile (ALS), dando luogo a forme binarie e libere che mostrano maggiore diffusione extra-vascolare e disponibilità per le cellule bersaglio. Inoltre, la soppressione ipofisaria della produzione di GH determina riduzione della sintesi di IGFBP-3 e di ALS, determinando ulteriore incremento delle forme libere o a più basso peso molecolare.
Poco è noto dei meccanismi molecolari che sostengono la iperproduzione di big IGF-2 da parte delle cellule tumorali. Alcuni studi sperimentali dimostrerebbero che le forme immature di big IGF-II sarebbero la conseguenza di un deficit cellulare dell’enzima pro-ormone convertasi 4 (17).
I criteri per la diagnosi di NICTH, proposti nel 1990 da Teale e Marks, includono valori inappropriatamente elevati di IGF-II associati a basse concentrazioni di IGF-I (rapporto concentrazioni IGF-II/IGF-I > 3)(18). Inoltre, si associano ridotte concentrazioni di insulina, peptide C e pro-insulina e soppressione di produzione di GH. La dimostrazione di elevati livelli di big IGF-II è patognomonica, ma non sono disponibili metodiche immunometriche di pronto utilizzo per tale determinazione, che necessita di più complesse tecniche di tipo cromatografico.
L’intervento terapeutico nei pazienti affetti da NICTH è complesso, multimodale e spesso frustrante (1,19,20). Nei pazienti portatori di SFT benigni l’asportazione della neoplasia determina la pronta risoluzione dell’ipoglicemia. Al contrario, la NICTH associata a tumori maligni in fase avanzata può essere affrontata attraverso l’adozione di:
- terapie anti-neoplastiche specificamente rivolte alla neoplasia responsabile del quadro sindromico;
- terapie di supporto finalizzate al mantenimento di adeguati valori glicemici (somministrazione ripetute di glucosio per os o per via infusionale);
- glucocorticoidi, che sembrano rappresentare l’approccio più efficace e in grado di garantire un miglior controllo nel tempo della glicemia. Viene fatto ricorso a dosi progressivamente crescenti fino ad arrivare a dosi equivalenti di prednisolone di 3 mg/kg/die;
- ormone della crescita (rhGH), in grado di stimolare la produzione di IGFB-3 e ALS e di aumentare la gluconeogenesi epatica. Può essere associato ai glucocorticoidi;
- approcci sperimentali. Considerando la patogenesi della NICHT, è prevedibile che l’utilizzo di inibitori molecolari della via di trasmissione del segnale innescata da IGF1-R possa portare a un miglioramento del quadro metabolico. Sono in fase di valutazione clinica diversi anticorpi in grado bloccare l’attività di IGF1-R. Tuttavia, nessuno di questi composti è stato utilizzato nel controllo della NICTH.
Bibliografia
- de Groot J, Rikhof B, van Doorn J, et al.Non-islet cell tumour-induced hypoglycaemia: a review of the literature including two new cases. Endocr Relat Cancer 2007, 14: 979–93.
- Doege KW. Fibrosarcoma of the mediastinum. Ann Surg 1930, 92: 955-60.
- Potter RE. Intrathoracic tumors. Radiology 1930, 14: 60-2.
- Kalebi A, Hale M, Wong M, et al. Surgically cured hypoglycemia secondary to pleural solitary fibrous tumour: case report and update review on the Doege-Potter syndrome. J Cardiothorac Surg 2009, 4: 45.
- Schutt R, Gordon T, Bhabhra R, et al. Doege-Potter syndrome presenting with hypoinsulinemic hypoglycemia in a patient with a malignant extrapleural solitary fibrous tumor: a case report. J Med Case Rep 2013, 7: 11.
- Tominaga N, Kawarasaki C, Kanemoto K et al. Recurrent solitary fibrous tumor of the pleura with malignant transformation and non-islet cell tumor-induced hypoglycemia due to paraneoplastic overexpression and secretion of high-molecular-weight Insulin-like Growth Factor II. Intern Med 2012, 51: 3267-72.
- Famà F, Le Bouc G, Barrande A, Villeneuve MGB. Solitary fibrous tumour of the liver with IGF-II-related hypoglycaemia. A case report. Langenbecks Arch Surg 2008, 393: 611-6.
- Nadler WA, Wolfer JH. Hepatogenic hypoglycemia associated with primary liver cell carcinoma. Arch Int Med 1929, 44: 700–5.
- Samlani-Sebbane Z, Azeddine Diffaa A, Khadija Krati K, et al. Fatal hypoglycaemia from IGF II hyperproduction as a complication of a mesenteric gastrointestinal stromal tumour. Arab J Gastroenterol 2011, 12: 171-2.
- Barra W, Castro G, Oliveira A, et al. Symptomatic hypoglycemia related to inappropriately high IGF-II serum levels in a patient with desmoplastic small round cell tumor. Case Rep Med 2010, 2010: 684045.
- Powter L, Phillips L, Husbands E. A case report of non-islet cell tumour hypoglycaemia associated with ovarian germ-cell tumour. Palliat Med 2013, 27: 281-3.
- Morioka T, Ohba K, Morita H, et al. Non–islet cell tumor-induced hypoglycemia associated with macronodular pulmonary metastases from poorly differentiated thyroid carcinoma. Thyroid 2013 Sep 11 (Epub ahead of print).
- Ida T, Morohashi T, Ohara H, et al. Gastric neuroendocrine carcinoma with non-islet cell tumor hypoglycemia associated with enhanced production of Insulin-like Growth Factor II. Intern Med 2013, 52: 757-60.
- Shibata Y, Ito Y, Fujita H, et al. A novel case of functional gastric neuroendocrine carcinoma occurred after endoscopic submucosal dissection. Case Rep Gastrointest Med 2013, 2013: 148761.
- Rikhof B, van Doorn J, Suurmeijer A, et al. Insulin-like growth factors and insulin-like growth factor-binding proteins in relation to disease status and incidence of hypoglycaemia in patients with a gastrointestinal stromal tumour. Ann Oncol 2009, 20: 1582-8.
- Qiu Q, Yan X, Bell M, et al. Mature IGF-II prevents the formation of ‘‘big” IGF-II/IGFBP-2 complex in the human circulation. Growth Horm IGF Res 2010, 20: 110-7.
- Tani Y, Tateno T, Izumiyama H, et al. Defective expression of prohormone convertase 4 and enhanced expression of insulin-like growth factor II by pleural solitary fibrous tumor causing hypoglycemia. Endocr J 2008, 55: 905-11.
- Teale JD, Marks V. Inappropriately elevated plasma insulin-like growth factor II in relation to suppressed insulin-like growth factor I in the diagnosis of non-islet cell tumour hypoglycaemia. Clin Endocrinol 1990, 33: 87–98.
- Davda R, Seddon BM. Mechanisms and management of non-islet cell tumour hypoglycaemia in gastrointestinal stromal tumour: case report and a review of published studies. Clin Oncol (R Coll Radiol) 2007, 19: 265-8.
- Okushina K, Asaokaa Y, Fukudab I, et al. IGF-II producing hepatocellular carcinoma treated with Sorafenib: metabolic complications and a foresight to molecular targeting therapy to the IGF signal. Case Rep Gastroenterol 2012, 6: 784-9.
Osteomalacia oncogenica
Alfredo Scillitani
Endocrinologia, Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG)
Fisiopatologia ed eziopatogenesi
L’osteomalacia tumorale (TIO, Tumor-Induced Osteomalacia) è una sindrome osteo-metabolica paraneoplastica rara, di cui sono stati descritti finora meno di 400 casi (1,4).
È dovuta alla secrezione da parte di piccoli tumori mesenchimali di FGF-23. Questo è un ormone che legandosi fisiologicamente al proprio recettore e interagendo con altri cofattori (Klotho), riduce trascrizione ed espressione dei cotrasportatori sodio-fosfato 2a e 2c. L’effetto finale è quello di diminuire il riassorbimento tubulare del fosforo, con conseguente iperfosfaturia e ipofosforemia. Inoltre, FGF-23 riduce l’espressione della 1α-idrossilasi renale e aumenta quella della 24-idrossilasi renale, favorendo così l’idrossilazione in posizione 24 della 25OH-vitaminaD, determinando alla fine una riduzione dei livelli di 1-25(OH)2vitaminaD. Tale riduzione è di fatto più importante, se si considera che l’ipofosforemia normalmente aumenta espressione e attività dell’1α-idrossilasi.
TIO è in genere dovuto a piccoli tumori mesenchimali a lenta crescita, con caratteristiche istologiche di benignità (solo nel 5% dei casi sono maligni con metastasi generalmente polmonari), che possono infiltrare il tessuto connettivo circostante. Originano dai tessuti molli (55% casi) o dall’osso (40% casi), sono localizzati alle estremità, al distretto testa/collo o alla mandibola (strange tumors in strange places). Dal punto di vista istologico, i più frequenti sono displasia fibrosa, emangiopericitoma, osteosarcoma, condroblastoma, fibroma condro-mixoide, istiocitoma fibroso maligno, tumore a cellule giganti ed emangioma (all’esame microscopico sono presenti cellule stellate o fusiformi con basso indice mitotico in matrice mixoide che può contenere osso lamellare/cartilagine/osteoide/cellule giganti). La possibile infiltrazione del tessuto circostante si traduce da un punto di vista pratico nella necessità di eseguire al momento dell’intervento un’ampia resezione dei margini chirurgici, per evitare la persistenza o la recidiva della malattia (3). La diagnosi istologica di malignità è difficile e infatti nelle rare metastasi le caratteristiche istologiche delle lesioni metastatiche sembrano benigne (3). Oossono recidivare anche dopo anni dalla chirurgia.
Il tumore produce fosfatonine (FGF-23 ma anche MEPE, SFRP-4, FGF-7), che causano ipofosfatemia attraverso 3 meccanismi:
- inibizione del riassorbimento tubulare del fosforo, con aumento di fosfaturia e ipofosfatemia;
- inibizione della sintesi di 1,25OHD3 con ridotto assorbimento intestinale di fosforo e calcio e conseguente iperparatiroidismo secondario;
- inibizione della mineralizzazione della matrice osteoide.
Da un punto di vista isto-morfometrico, tutto questo si traduce a livello osseo in una grave osteomalacia, con importante deficit di mineralizzazione e numerose fratture.
Clinica
La sintomatologia è aspecifica e peggiora progressivamente negli anni. Sono presenti dolori ossei, debolezza muscolare progressiva fino all’allettamento, fratture multiple; i pazienti pediatrici hanno un quadro di rachitismo con ritardo di crescita.
È stato riportato un tempo di 2.5–28 anni tra l’inizio dei sintomi e la diagnosi (2), mentre sono in media necessari 5 anni per l’identificazione della neoplasia dopo la diagnosi (1).
La malattia deve essere sospettata per la presenza della sintomatologia associata a ipofosforemia.
Diagnosi
Da un punto di vista biochimico si osservano ipofosforemia, iperfosfaturia (ridotto TmPO4/GFR) e valori bassi o inappropriatamente normali in rapporto all’ipofosforemia di 1-25(OH)2vitaminaD, aumentati livelli di fosfatasi alcalina (indice di aumentato rimodellamento scheletrico), mentre PTH e calcemia sono nella norma.
Dopo aver confermato l’ipofosforemia, deve essere calcolato il TmPO4/GFR, cioè la capacità di riassorbimento massimale tubulare del fosfato, che, se risulta basso, indica che la perdita renale di fosfati è la causa dell’ipofosforemia. Il TmPO4/GFR, si determina misurando la fosforemia e creatininemia sierica e la fosfaturia e creatininuria a digiuno sulla 2° minzione del mattino; per il calcolo possono essere utilizzati vari algoritmi presenti sul web (3).
A questo punto un ulteriore supporto alla diagnosi può essere la misurazione dei livelli serici di 1-25(OH)2vitaminaD, che risulteranno bassi o inappropriatamente normali (in riferimento all’ipofosforemia).
Infine, può essere dosato il livello plasmatico di FGF23, che risulterà molto elevato e confermerà la diagnosi di malattia di perdita di fosfato FGF23-dipendente.
La diagnosi differenziale deve considerare essenzialmente alcune malattie genetiche, che sono delle fenocopie, e per potersi orientare può essere importante considerare l’età di inizio della sintomatologia, la storia familiare, possibili patologie dentarie. Generalmente più giovane è il paziente, più probabile è la causa genetica. Per un’accurata diagnosi differenziale si rimanda alla letteratura (3). Ci sono peraltro cause acquisite che possono determinare un quadro clinico e biochimico sostanzialmente sovrapponibile a quello del TIO. Tra esse ricordiamo le tubulopatie da metalli pesanti e da farmaci (i.e. da tenofovir, aminoglicosidici). In queste ultime forme acquisite i livelli di FGF23 sono bassi.
Localizzazione del tumore
Il tumore può essere presente in qualsiasi parte del corpo, dalla testa ai piedi, ed è spesso localizzato nelle estremità. Questa informazione è importante, perché testa, gambe e piedi, braccia e mani devono essere sempre valutate nei vari esami eseguiti per la localizzazione della neoplasia. Pertanto, un’accuratissima visita del paziente, con particolare osservazione di qualsiasi minima lesione presente nel sottocute dalla testa ai piedi, potrebbe già essere di aiuto per la localizzazione. Metodiche funzionali utili sono:
- 18F-FDG-PET/TC che è una metodica sensibile;
- scintigrafia con Octreoscan (recettori della somatostatina sono stati identificati su molti TIO) eventualmente combinata con metodica TC;
- 68Ga-DOTANOC PET/TC, che combinerebbe la specificità dell’octreotide con la sensibilità della PET/TC.
In queste metodiche “funzionali” alcune aree del corpo non sono ben visualizzate: l’encefalo da 18F-FDG-PET; fegato e milza da octreotide. Se anche dopo aver sottoposto il paziente a tali indagini la neoplasia non è stata identificata, è indicata l’esecuzione di altri esami come TC o RM (3,4).
Talora è stato eseguito un campionamento venoso con dosaggio di FGF23, per confermare che una lesione evidenziata con altri esami è responsabile della sindrome (1).
Nonostante tutte le indagini eseguite, il tumore potrebbe non essere localizzato. In tale evenienza è opportuno ripetere gli esami di localizzazione a distanza (dopo un anno).
Terapia e follow-up
Il trattamento risolutivo è l’asportazione chirurgica della lesione con ampio margine di resezione, che determina una rapida normalizzazione del quadro biochimico e quindi una rimineralizzazione dell’osso.
Comunque, se dopo aver posto la diagnosi la neoplasia non è stata individuata o, se individuata, non è stata asportata del tutto, è indicata la terapia medica con fosfato (1-3 g/die, suddivisi in 3-4 somministrazioni) e calcitriolo (1-3 µg/die). La terapia medica deve essere individualizzata, per migliorare i sintomi e cercare di normalizzare la fosfatasi alcalina. Tale terapia riduce il dolore osseo e muscolare e favorisce la mineralizzazione.
È in corso di studio l’anticorpo monoclonale anti-FGF23.
Bisogna monitorare il quadro biochimico per prevenire gli effetti negativi della terapia (ipercalcemia, calcolosi renale e nefrolitiasi). Pertanto, è necessario misurare calcio serico ed urinario, funzione renale e PTH, inizialmente ogni mese, quindi ogni 3 mesi. È riportata un’aumentata incidenza di iperparatiroidismo dopo prolungato trattamento con fosforo. In pazienti intolleranti a tale terapia, sono stati descritti casi singoli trattati con cinacalcet e octreotide (2).
Bibliografia
- Ruppe MD, Jane de Beur SM. Disorders of Phosphate Homeostasis. In: Rosen CJ, et al, Editors; Primer on the metabolic bone diseases and disorders of mineral metabolism; 8th Edition, Wiley-Blackwell 2013: 601-12.
- Chiam P, Tan HC, Bee YM, Chandran M. Oncogenic osteomalacia – Hypophosphatemic spectrum from “benignancy” to “malignancy”. Bone 2013, 53: 182-7.
- Chong WH, Molinolo AA, Chen CC, Collins MT. Tumor-induced osteomalacia. Endocr Relat Cancer 2011, 18: R53-77.
- Jiang Y, Xia WB, Xing XP, et al. Tumor-induced osteomalacia: an important cause of adult-onset hypophosphatemic osteomalacia in China: report of 39 cases and review of the literature. J Bone Miner Res 2012, 27: 1967-75.
Complicanze endocrine delle patologie oncologiche, della target therapy e nei cancer survivors
I primi studi sui pazienti affetti da malattia neoplastica il cui trattamento aveva dato una lunga sopravvivenza (per questo chiamati "cancer survivors") sono iniziati nel 1986 a seguito del primo Congresso della National Coalition for Cancer Survivorship che si è svolto a Albuquerque nel New Mexico. Risalgono però a numerosi anni dopo, dal 1997 in poi, i primi studi sistematici longitudinali sugli effetti collaterali delle terapie anti-neoplastiche nei bambini sopravvissuti. Le possibili sequele dei pregressi trattamenti anti-neoplastici, si presentavano a carico di numerosi organi e apparati: in particolare le prime descrizioni a carico del sistema endocrino sono state a carico dell'asse ipotalamo-ipofisario e delle ghiandole corticosurrenaliche.