Terapia farmacologica per i NET duodeno-pancreatici nell'ambito della MEN-1: quando e quale?

Maria Vittoria Davì1 & Gabriele Luppi2
1UOC Medicina Interna D, Policlinico GB Rossi, AOUI Verona
2Oncologia Medica, Azienda Ospedaliero-Universitaria di Modena
Allo stato attuale non vi sono studi pubblicati focalizzati sul trattamento dei NET nell’ambito delle MEN-1. Le recenti linee-guida dell’ENETS si riferiscono ai NET in generale, pertanto non vi sono al momento motivi per considerare il trattamento dei NET MEN-1 correlati diverso da quello della controparte sporadica.
La terapia medica con gli analoghi della somatostatina a lunga durata d'azione (octreotide e lanreotide) dei GEP-NET associati a MEN-1 ha le stesse indicazioni per le forme corrispettive sporadiche, ovvero per il controllo della sindrome ormonale delle forme funzionanti e per il controllo della crescita tumorale delle forme ben differenziate. Non vi sono evidenze specifiche che riguardino i NET MEN-1 correlati, per cui in questo paragrafo si farà riferimento alla letteratura e alle linee guida dei NET in generale.
Per quanto riguarda il controllo della sindrome ormonale, octreotide e lanretotide ottengono una risposta media intorno al 73% (range 50-100%)(1). Pertanto possono essere utilizzati nei NET pancreatici funzionanti associati alla MEN-1, in particolare VIPomi, glucagonomi non suscettibili di chirurgia o metastatici, mentre meno efficaci sono nel controllo della sindrome ipoglicemica degli insulinomi (2).
Il controllo della crescita nei NET pancreatici si basa prevalentemente su studi retrospettivi o prospettici non randomizzati, riguardanti oltre 500 pazienti, con una stabilizzazione di malattia intorno al 50% (1-4). In tale caso la terapia con gli analoghi della somatostatina rappresenta l'opzione iniziale nelle forme ben differenziate (G1), previa verifica della presenza di recettori 2 per la somatostatina mediante Octreoscan o 68Ga-DOTATOC-PET. Tuttavia, nei NET pancreatici G1-G2 dovrebbero essere prese in considerazione altre opzioni terapeutiche, come chemioterapia, sunitinib, everolimus, terapia radio-recettoriale, anche se attualmente mancano evidenze su quale sia la sequenza ideale (5). L’alfa-interferone non aggiunge un vantaggio terapeutico quando somministrato in combinazione con gli analoghi della somatostatina rispetto agli analoghi usati da soli, come dimostrato in 2 studi randomizzati (6,7).
Gli elementi che influiscono maggiormente sulla decisione terapeutica sono l’istologia ed il grado tumorale, la sede del tumore primitivo, l’eventuale produzione di ormoni, la presenza o meno di sintomi correlati al tumore, l’espressione di recettori della somatostatina, la presenza di malattia extra-epatica (8).
Tradizionalmente gli agenti chemioterapici più efficaci in questi tipi di tumori sono Streptozotocina in combinazione con 5-fluorouracile, Doxorubicina e in parte anche Dacarbazina. Temozolomide, Oxaliplatino e Capecitabina, in monoterapia o in regimi di combinazione, hanno dimostrato una certa efficacia in studi di fase II. I pazienti che beneficiano maggiormente di un trattamento anti-blastico sono quelli con tumori scarsamente differenziati che, indipendentemente dalla sede di origine, sono spesso candidati a un trattamento chemioterapico di prima linea con Cisplatino ed Etoposide, con tassi di risposta del 55-80% e durata mediana di risposta di 8-11 mesi (9).
Everolimus, un inibitore orale di mTOR, ha dimostrato attività nei GEP-NET. Lo studio RADIANT-3, prospettico, randomizzato di fase III, confrontava everolimus 10 mg/die in associazione alla migliore terapia di supporto versus la migliore terapia di supporto in pazienti con pNET ben differenziati avanzati in progressione. Tale studio ha dimostrato un vantaggio di 6.4 mesi per la sopravvivenza libera da progressione di malattia nel gruppo dei pazienti trattati con everolimus, portando quindi alla registrazione del farmaco in questa tipologia di pazienti (10).
Sunitinib malato è un inibitore tirosin-chinasico multi-target, che in uno studio randomizzato, placebo-controllato, di fase III in pazienti con pNET ben differenziati avanzati ha mostrato un prolungamento della sopravvivenza mediana libera da progressione di malatia (11.4 mesi vs 5.4 mesi). Questo studio ha portato pertanto le autorità regolatorie all’approvazione di questo farmaco in pazienti con pNET avanzati ben differenziati in progressione (11).
La terapia medica della sindrome di Zollinger–Ellison (ZES) associata a MEN-1 è sovrapponibile a quella delle forme sporadiche e prevede l’uso di inibitori di pompa protonica (PPI) ad alte dosi (per es. omeprazolo 60 mg/die) per il controllo della secrezione acida. Gli analoghi della somatostatina non sono in genere raccomandati per il controllo dell’acidità gastrica, ma solo nei casi resistenti alla sola terapia con PPI (4). Possono essere utilizzati allo scopo di ottenere un effetto anti-proliferativo nelle forme metastatiche (2). Per quanto riguarda i dati della letteratura sull’argomento, non vi sono evidenze specifiche sulle ZES/MEN-1, ma solo sulla ZES sporadica che riguardano casistiche poco numerose, data anche la rarità della patologia. In uno studio su 15 pazienti affetti da gastrinoma maligno con progressione epatica si è ottenuta la stabilizzazione di malattia nel 47% e la riduzione nel 6% in corso di trattamento con octreotide LAR 20-30 mg/mese, con una durata media di risposta di 25 ± 6 mesi (12). La stabilizzazione di malattia si è ottenuta anche in un altro gruppo di 11 gastrinomi associati a MEN-1, di cui 63.6% trattati con analoghi della somatostatina e 27.3% con chemioterapia (13). Lanreotide sembra avere un effetto simile ad octreotide nel controllo della sindrome clinica e stabilizzazione di malattia dei gastrinomi (14,15).
Per quanto riguarda i carcinoidi gastrici di tipo 2 associati alla ZES/MEN-1, attualmente le linee guida dell’ENETs non consigliano il trattamento con analoghi, anche se vi sono segnalazioni in letteratura di una regressione dopo trattamento con octreotide LAR (16,17).
Bibliografia
- Oberg KE, Reubi JC, Kwekkeboom DJ, Krenning EP. Role of somatostatins in gastroentero-pancreatic neuroendocrine tumor development and therapy. Gastroenterology 2010, 139: 742-53.
- Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012, 97: 2990-3011.
- Modlin IM, Pavel M, Kidd M, et al. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther 2010, 31: 169-88.
- Jensen RT, Cadiot G, Brandi ML, et al. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms: functional pancreatic endocrine tumor syndromes. Neuroendocrinology 2012, 95: 98-119.
- Pavel M, Baudin E, Couvelard A, et al. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology 2012, 95: 157-76.
- Arnold R, Rinke A, Klose KJ, et al. Octreotide versus octreotide plus interferon in endocrine gastroenteropancreatic tumors: a randomized trial. Clin Gastroenterol Hepatol 2005, 3: 761-71.
- Faiss S, Pape UF, Bohmig M, et al. International Lanreotide and Interferon Alfa Study Group: prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors – the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol 2003, 21: 2689-96.
- Pavel M, Kidd M, Modlin I. Systemic therapeutic options for carcinoids. Semin Oncol 2013, 40: 84-99.
- Fjällskog ML, Granberg DP, Welin SL, et al. Treatment with cisplatin and etoposide in patients with neuroendocrine tumors. Cancer 2001, 92: 1101-7.
- Yao JC, Shah MH, Ito T, et al. RAD001 in Advanced Neuroendocrine Tumors, Third Trial (RADIANT-3) Study Group. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011, 364: 514-23.
- Raymond E, Dahan L, Raoul JL, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Eng J Med 2011, 364: 501-13.
- Shojamanesh H, Gibril F, Louie A, et al. Prospective study of the antitumor efficacy of long-term octreotide treatment in patients with progressive metastatic gastrinoma. Cancer 2002, 94: 331-43.
- Nikou GC, Toubanakis C, Nikolaou P, et al. Gastrinomas associated with MEN-1 syndrome: new insights for the diagnosis and management in a series of 11 patients. Hepatogastroenterology 2005, 52: 1668-76.
- Gaztambide S, Vazquez JA. Short- and long-term effect of a long-acting somatostatin analogue, lanreotide (SR-L) on metastatic gastrinoma. J Endocrinol Invest 1999, 22: 144-6.
- Tomassetti P, Migliori M, Gullo L. Slow-release lanreotide treatment in endocrine gastrointestinal tumors. Am J Gastroenterol 1998, 93: 1468-71.
- Delle Fave G, Kwekkeboom DJ, Van Cutsem E, et al. ENETS Consensus Guidelines for the management of patients with gastroduodenal neoplasms. Neuroendocrinology 2012, 95: 74-87.
- Tomassetti P, Migliori M, Caletti GC, et al. Treatment of type II gastric carcinoid tumors with somatostatin analogues. N Engl J Med 2000, 343: 551-4.
Adenomi ipofisari nell'ambito della MEN-1
Roberto Attanasio
Endocrinologia, Istituto Galeazzi, Milano
Epidemiologia
Adenomi ipofisari compaiono nel 30-40% dei pazienti con MEN-1 (1). La prevalenza di lesioni ipofisarie sarebbe maggiore nei casi di MEN 1 sporadici rispetto ai casi familiari (59% vs 34%)(2).
Gli adenomi possono essere la prima manifestazione clinica di MEN-1: questo si verificherebbe nel 17% in media (2), in particolare nel 25% dei casi sporadici di MEN e nel 10% dei casi familiari (3).
Il ritardo fra la prima e la seconda manifestazione di MEN-1 è più lungo quando la lesione ipofisaria è la prima a comparire: 9 anni vs 4-5 anni quando la prima manifestazione è iperparatiroidismo o NET (2). Non è noto il motivo di tale differenza (si pensa meno a questa diagnosi in un paziente che si presenta con adenoma ipofisario?).
Nel 2% dei casi gli adenomi ipofisari possono essere l'unica manifestazione di MEN-1 (diagnosi genetica, 2).
La lesione ipofisaria può comparire a qualunque età: come estremi, sono riportati un caso diagnosticato a 5 anni (4) e uno a 83 anni (2).
E' stata riportata la presenza di tutti i sottotipi di adenomi, con una distribuzione non molto diversa da quella degli adenomi sporadici (2,5): PRLomi 62%, GHomi 9%, ACTHomi 6%, adenomi misti 10%, NFPA 15%. Sono stati riportati anche casi di ipersecrezione biochimica e clinica di gonadotropine (con sindrome da iperstimolazione ovarica) e carcinomi (6).
Quali sono le caratteristiche degli adenomi ipofisari nell'ambito della MEN-1?
I dati più importanti derivano dal network franco-belga (2). In confronto ai casi sporadici, gli adenomi associati a MEN-1 sono più spesso pluri-ormonali (39% vs 22%), adenomi doppi (4% vs 0.1%), associati a iperplasia del tessuto peri-tumorale (4% vs 0%)(7,8). Non c'è invece differenza di rapporto fra forme ipersecernenti e non funzionanti, nè di caratteristiche proliferative (Ki67 o indice mitotico, pur con i limiti di tali tecniche nel prevedere il comportamento biologico)(8).
Dal punto di vista della gestione clinica, gli adenomi associati a MEN-1 sono più voluminosi (macroadenomi 85% vs 64%), più aggressivi (con maggiore invasività istologica- 30% vs 10% - ma non radiologica) e più resistenti al trattamento (> 45% vs 10-40% di persistenza di ipersecrezione dopo trattamento multimodale) (9).
Come monitorare per la comparsa di adenoma ipofisario un portatore noto di mutazione MEN-1
È opportuno eseguire:
- ogni anno dosaggio di PRL e IGF-I;
- ogni 3-5 anni dosaggio di ACTH e CLU insieme a RM della regione ipofisaria (10,11).
Nel caso di anomalie biochimiche o radiologiche, passare a una diagnostica di secondo livello, per confermare/escludere ipersecrezioni o ipopituitarismo.
Come trattare gli adenomi ipofisari nel contesto della MEN-1
Il trattamento è identico a quello dei corrispondenti adenomi ipofisari sporadici (11). E' importante ricordare però che anche nei pazienti apparentemente "guariti" con l'intervento neurochirurgico (dove questo sia indicato), lo screening deve continuare indefinitamente, analogamente alle forme sporadiche, perchè dal tessuto ipofisario residuo possono nascere nuovi tumori.
L'ampia casistica franco-belga dimostra che a parità di condizioni di partenza e di trattamento gli adenomi nell'ambito della MEN-1 hanno peggiore risposta alle terapie (normalizzazione ormonale nel 42% vs 90% degli adenomi non MEN) che insieme alla maggior percentuale di macroadenomi indica una maggiore aggressività (2, 12).
E' quindi necessario un follow-up molto attento. Anche se non ci sono dati che indichino che l'albero decisionale sulle tappe terapeutiche debba essere diverso da quello impiegato negli adenomi non MEN, è probabile che sia più frequente la necessità di terapie più aggressive (12).
Come indagare l'esistenza di MEN-1 in un paziente che si presenta con adenoma ipofisario?
La mutazione MEN-1 si trova nello 0.6% degli adenomi sporadici (8).
E' sempre opportuna un'accurata anamnesi familiare, in tutti va eseguito un dosaggio di calcemia e nei pazienti in cui l'adenoma si presenta prima dei 20 anni lo screening deve essere più accurato (13).
E' stato descritto un algoritmo più raffinato (figura) per tentare di predire l'esito del test genetico per la mutazione MEN-1 nei portatori di tumori endocrini apparentemente sporadici, che mette insieme l'età, la familiarità e il tipo di tumore. In quell'algoritmo comunque i tumori ipofisari hanno il valore predittivo più scarso (14).
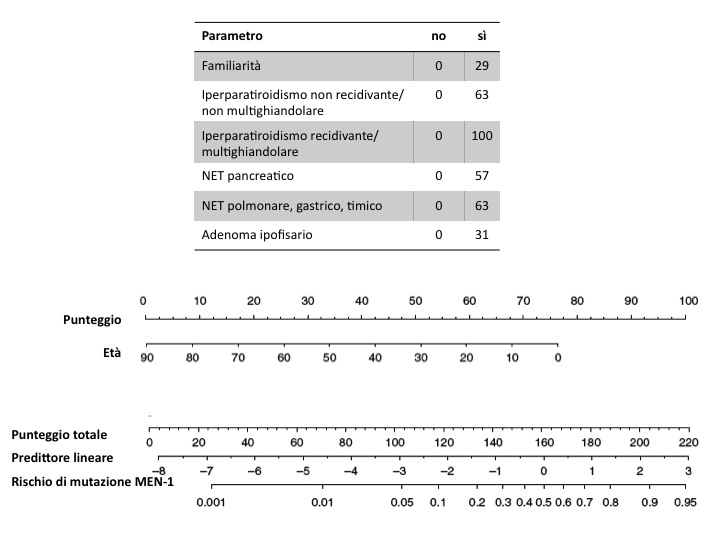
Nomogramma per calcolare il rischio di mutazione MEN-1 (modificato da 14)
Si procede in 3 fasi:
1. Sommare i punteggi relativi ai parametri elencati nella tabella in alto
2. Aggiungere il punteggio relativo all’età (al centro)
3. Segnare la somma ottenuta sul regolo in basso, in corrispondenza del punteggio totale e abbassare una perpendicolare fino a incrociare la scala logaritmica in basso.
Esempio: paziente di 50 anni, senza familiarità (0) con iperpara non recidivante (63) e NET pancreatico (57); al punteggio di 120 (63+57) si somma quello relativo all’età (33); al totale di 153 corrisponde il 38% di probabilità di mutazione
Bibliografia
- Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012, 97: 2990-3011.
- Verges B, Boureille F, Goudet P, et al. Pituitary disease in MEN type 1 (MEN1): data from the France-Belgium MEN1 multicenter study. J Clin Endocrinol Metab 2002, 87: 457-65.
- Falchetti A, Marini F, Luzi E, et al. Multiple endocrine neoplasia type 1 (MEN1): not only inherited endocrine tumors. Genet Med 2009, 11: 825-35.
- Stratakis CA, Schussheim DH, Freedman SM, et al. Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2000, 85: 4776-80.
- O'Brien T, O'Riordan D, Gharib H. et al. Results of treatment of pituitary disease in multiple endocrine neoplasia, type I. Neurosurgery 1996, 39: 273-9.
- Benito M, Asa SL, LiVolsi VA, et al. Gonadotroph tumor associated with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2005, 90: 570-4.
- Capella C, Riva C, Leutner M, La Rosa S. Pituitary lesions in multiple endocrine neoplasia syndrome (MENS) Type 1. Pathol Res Pract 1995, 191: 345-7.
- Trouillas J, Labat-Moleur F, Sturm N, et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol 2008, 32: 534-43.
- Syro LV, Scheithauer BW, Kovacs K, et al. Pituitary tumors in patients with MEN1 syndrome. Clinics 2012, 67(S1): 43-8.
- Burgess JR, Shepherd JJ, Parameswaran V, et al. Spectrum of pituitary disease in multiple endocrine neoplasia type 1 (MEN 1): clinical, biochemical, and radiological features of pituitary disease in a large MEN 1 kindred. J Clin Endocrinol Metab 1996, 81: 2642-6.
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001, 86: 5658-71.
- Beckers A, Betea D, Valdes Socin H, Stevenaert A. The treatment of sporadic versus MEN1-related pituitary adenomas. J Int Med 2003, 253: 599-605.
- Goudet P, Bonithon-Kopp C, Murat A, et al. Gender-related differences in MEN1 lesion occurrence and diagnosis: a cohort study of 734 cases from the Groupe d'etude des Tumeurs Endocrines. Eur J Endocrinol 2011, 165: 97-105.
- de Laat JM, Tham E, Pieterman CRC. Predicting the risk of multiple endocrine neoplasia type 1 for patients with commonly occurring endocrine tumors. Eur J Endocrinol 2012, 167: 181-7.
- Nunes VS, Souza GL, Perone D, et al. Frequency of multiple endocrine neoplasia type 1 in a group of patients with pituitary adenoma: genetic study and familial screening. Pituitary 2013 DOI 10.1007/s11102-013-0462-8.
Neoplasie surrenaliche nell'ambito della MEN-1
Anna Pia
Dipartimento di Scienze Cliniche e Biologiche, Medicina Interna 1 a Indirizzo Endocrinologico, AOU San Luigi di Orbassano, Università di Torino
Epidemiologia
La prevalenza di tumori asintomatici della corticale del surrene nei pazienti con MEN-1 è di circa il 40%, con percentuali che variano dal 20 al 73% nelle diverse casistiche (1,2). Questa estrema variabilità dipende essenzialmente dalle tecniche radiologiche di screening utilizzate. L’interessamento della ghiandola surrenalica nella MEN-1 è infatti frequente con l’utilizzo di TAC e/o RMM di ultima generazione e, ancor di più, con l’eco-endoscopia. Quest’ultima tecnica, usata nella MEN-1 anche per lo screening di lesioni pancreatiche, può evidenziare piccole lesioni surrenaliche e/o lievi ingrandimenti del surrene nel 55-73% dei casi (3,4). Il significato clinico/patologico di queste lesioni surrenaliche non è noto, ma, come descritto per gli incidentalomi di piccole dimensioni e asintomatici, nessuna lesione < 10 mm sembrerebbe evolutiva e/o associata a disfunzione endocrina (2,3).
Va segnalato che le discrepanze tra le percentuali di tumori surrenalici riportate in letteratura deriva, oltre che dalle diverse tecniche radiologiche utilizzate, anche dalle diverse modalità di definizione delle lesioni surrenaliche riscontrate. In alcune casistiche, infatti, vengono assimilate ai “tumori surrenalici” lesioni surrenaliche di diversa natura e pertanto vengono inclusi adenomi corticali, iperplasia, adenomi multipli ed iperplasia nodulare, cisti e carcinomi (1).
Nel maggiore studio multicentrico sinora pubblicato sull’interessamento del surrene nell’ambito della MEN1 (715 casi), circa 20.4% dei pazienti presentavano “ingrandimenti surrenalici”, prevalentemente unilaterali, ma solo nel 10% dei casi si trattava di tumori corticosurrenalici > 10 mm. Limite dello studio è la raccolta della casistica in un arco di 40 anni, con impiego di tecniche radiologiche eterogenee e mancanza di una revisione centralizzata delle immagini, con possibile sottostima dei tumori (2).
Manifestazioni cliniche e diagnosi
La maggior parte dei tumori surrenalici nel contesto di MEN-1 sono adenomi della corticale del surrene, non secernenti e asintomatici. Un’ipersecrezione ormonale è descritta in meno del 10% dei pazienti: si tratta principalmente di casi di ipercortisolismo ACTH-indipendente e iperaldosteronismo primitivo, mentre è rara l’associazione con feocromocitoma (circa 1% dei casi), a differenza di quanto avviene nell’incidentaloma surrenalico (2, 5). Molto rara è poi la secrezione di androgeni, isolata o associata ad ipercortisolismo, descritta unicamente in pazienti con neoplasia maligna del corticosurrene (2,6). Da segnalare che nelle casistiche della MEN-1 non viene preso in considerazione l’ipercortisolismo subclinico e il feocromocitoma potrebbe essere sottostimato, in quanto numerose casistiche non utilizzano le metanefrine nello screening dei casi silenti (2). I tumori surrenalici, specie se secernenti, possono anche essere la prima manifestazione di una MEN-1 (2, 7).
Le recenti linee guida sulla MEN-1 consigliano accertamenti biochimici (es. PRA/aldosterone, test di soppressione con desametasone a basse dosi, metanefrine/catecolamine) per i pazienti con segni/sintomi suggestivi per ipersecrezione surrenalica e per i pazienti con tumore surrenalico > 1 cm (1).
Importante è considerare il potenziale maligno dei tumori surrenalici MEN1-correlati.Nei pazienti affetti da MEN-1 la prevalenza del carcinoma del corticosurrene è bassa, circa dell’’1%, ma può superare il 13% in quelli con tumore surrenalico > 1 cm (1,2). Tale frequenza è significativamente superiore a quella riscontrata nelle lesioni surrenaliche di riscontro occasionale, come riportato da una recente review (8). Attualmente, in letteratura sono stati segnalati 16 di casi di MEN-1 associata a carcinoma del corticosurrene (2, 8); in 3 casi (18.7%) la neoplasia era bilaterale (2, 6).
Secondo alcuni autori, nella maggior parte dei casi di MEN-1 il carcinoma del surrene sarebbe diagnosticato in uno stadio iniziale (stadio I-II secondo classificazione ENS@T), avrebbe una crescita più lenta e un’aggressività minore rispetto alla forma sporadica, quasi a suggerire un’entità specifica (2). Certamente il programma di screening delle neoplasie in ambito della MEN-1, con imaging addominale periodico, facilita la diagnosi precoce e permette il trattamento chirurgico radicale, importantissimo fattore prognostico ai fini della sopravvivenza
Per quanto riguarda l’associazione tra lesioni surrenaliche e genotipo, i dati in letteratura sono al momento discordanti. Alcuni lavori hanno infatti segnalato una correlazione tra la presenza di lesioni surrenaliche e mutazioni nell’esone 2 e 10 nel gene della MEN-1 (6, 9), ma il dato non ha trovato riscontro in casistiche più numerose (2-4).
Terapia e follow-up
La gestione clinica dei tumori surrenalici nell’ambito di una MEN-1 dipende dalle caratteristiche radiologiche e dall’attività ormonale della lesione surrenalica.
Analogamente all’incidentaloma surrenalico, le dimensioni e le caratteristiche radiologiche del tumore, in particolare la densità espressa in unità Hounsfield alla TAC, possono orientare sulla natura della lesione surrenalica e condizionare il trattamento (5). Sulla terapia dei tumori surrenalici associati a MEN-1 non c’è tuttavia un consenso unanime, poiché per la maggior parte sono non funzionanti e benigni. Il rischio di malignità aumenta per le lesioni surrenaliche > 4 cm, ma nel caso di MEN-1 i tumori maligni possono avere dimensioni inferiori, comprese tra 1 e 4 cm. I tumori < 1 cm sono generalmente benigni e non evolutivi, per cui non è indicato un particolare follow-up.
Le recenti linee guida sulla MEN-1 consigliano pertanto l’intervento chirurgico di surrenectomia nei seguenti casi (1):
- tumori > 4 cm;
- tumori di dimensioni comprese tra 1-4 cm, se caratteristiche radiologiche atipiche/sospette per malignità;
- accrescimento significativo nell’arco di 6 mesi;
- tumori secernenti (analogamente alla terapia dei tumori non associati alla MEN-1).
Le lesioni surrenaliche di aspetto “benigno”, non secernenti e di diametro compreso tra 1 e 4 cm dovrebbero essere rivalutate con screening d’immagine (RMN o TAC), inizialmente a 6 mesi e poi con cadenza annuale (1). Se la lesione surrenalica resta dimensionalmente stabile, alcuni autori propongono di dilazionare il controllo d’immagine ogni 2 anni (3).
Non vi sono indicazioni precise in letteratura sul follow-up ormonale, da stabilire caso per caso in base a criteri clinici. In assenza di segni/sintomi clinici di ipersecrezione, i controlli potrebbero essere analoghi a quelli previsti per l’incidentaloma surrenalico: valutazione ormonale alla diagnosi e poi annuale per 5 anni (5).
Bibliografia
- Thakker RV, Newey PJ, Walls GV, et al. Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012, 97: 2990-3011.
- Gatta-Cherifi B, Chabre O, Murat A, et al. Adrenal involvement in MEN1. Analysis of 715 cases from the Groupe d'etude des Tumeurs Endocrines database. Eur J Endocrinol 2012, 166: 269-79.
- Schaefer S, Shipotko M, Meyer S, et al. Natural course of small adrenal lesions in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Eur J Endocrinol 2008, 158: 699-704.
- Waldmann J, Bartsch DK, Kann PH, et al. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg 2007, 392: 437-43.
- Terzolo M, Stigliano A, Chiodini I, et al. Italian Association of Clinical Endocrinologists. AME position statement on adrenal incidentaloma. Eur J Endocrinol 2011, 164: 851-70.
- Griniatsos JE, Dimitriou N, Zilos A, et al. Bilateral adrenocortical carcinoma in a patient with multiple endocrine neoplasia type 1 (MEN1) and a novel mutation in the MEN1 gene. World J Surg Oncol 2011, 9: 6.
- Alzahrani AS, Al-Khaldi N, Shi Y, et al. Diagnosis by serendipity: Cushing syndrome attributable to cortisol-producing adrenal adenoma as the initial manifestation of multiple endocrine neoplasia type 1 due to a rare splicing site MEN1 gene mutation. Endocr Pract 2008, 14: 595-602.
- Else T. Association of adrenocortical carcinoma with familial cancer susceptibility syndromes. Mol Cell Endocrinol 2012, 351: 66-70.
- Mazzuco TL, Durand J, Chapman A, et al. Genetic aspects of adrenocortical tumours and hyperplasias. Clin Endocrinol (Oxf) 2012, 77: 1-10.
Follow-up della MEN-1
Alberto Falchetti
Endocrinologo e Genetista Medico, Centro Hercolani, Bologna
Morbilità e mortalità nella MEN-1
Gli individui con sindrome MEN 1 hanno un aumento significativo del rischio di morte prematura, che giustifica la sorveglianza di quelli con mutazioni MEN-1 e/o una storia familiare di sindrome MEN-1 (1). Paradossalmente, una più lunga attesa di vita nella MEN-1 può comportare un aumento della morbilità e mortalità cumulativa per i tumori maligni associati a MEN-1, che rappresentano circa il 30% delle cause di decesso nella sindrome MEN-1 (2, 3).
È opportuna una “surveillance” di routine dei soggetti asintomatici portatori della mutazione germinale MEN-1 e di tutti gli altri soggetti a rischio per sviluppare i tumori associati a MEN-1 (es. coloro noti per essere affetti da MEN-1 e coloro con un genitore affetto e che non hanno eseguito il test genetico o in cui tale test non ha evidenziato alcuna mutazione nel gene MEN-1): test biochimici e d’immagine iniziando nella prima infanzia e continuando per tutta la vita.
Un precoce rilevamento e trattamento dei tumori neuroendocrini (NET) potenzialmente maligni, quali gastrinomi, carcinoidi timici e più raramente bronchiali (4-7), potrebbe ridurre la morbilità e mortalità legate a MEN-1. Un tale screening può identificare l’insorgenza della patologia circa dieci anni prima dello sviluppo dei sintomi, fornendo quindi l’opportunità per un trattamento precoce (8).
Valutazioni seguenti la diagnosi iniziale
Per stabilire l'entità della malattia in un individuo con diagnosi di MEN-1, si raccomanda che siano ricercati/valutati i seguenti tumori, più comunemente associati a MEN-1:
- Malattia multighiandolare delle paratiroidi;
- Gastrinoma e altri tumori neuroendocrini entero-pancreatici (funzionanti e non);
- Prolattinoma (si ricorda che sono stati descritti in letteratura, associati a MEN-1, anche tutti gli altri tipi di tumori ipofisari funzionanti)
Sorveglianza clinica, di minima, per soggetti noti avere la MEN-1 (test genetico positivo, membro familiare affetto, paziente indice affetto)
Indagini annuali
- Esame obiettivo (considerare la ricerca di tumori della pelle e lipomi associati a MEN-1) (a partire dai 5 anni)
- Calcemia totale (corretta per l'albumina) e/o calcio ionizzato (a partire da 8 anni d'età)
- Glicemia, insulinemia a digiuno, pro-insulina (a partire dai 5 anni)
- Gastrina a digiuno e/o stimolata (dai 20 anni)
- PRL, IGF-1 (dai 5 anni).
- Da prendere in considerazione: PTH intatto a digiuno.
Indagini d’immagine (da annuale a ogni 3-5 anni, in relazione all’evidenza biochimica di una neoplasia e/o dai segni e sintomi di un tumore MEN-1-correlato)
- TC o RM addominale (dall’età di 20 anni)
- RM cranio/sella turcica (dall’età di 5 anni)
- Da prendere in considerazione: TC annuale del torace, Octreoscan o se disponibile 68Ga-DOTA(TOC o NOC o TATE)-PET
Screening clinico per gli individui a rischio del 50% di avere la sindrome MEN-1, il cui status genetico è sconosciuto (parente di primo grado senza test genetico, membro di un gruppo clinico a rischio)
- Esame obiettivo (considerare la ricerca di tumori della pelle e lipomi associati a MEN-1)
- Test e consulenza genetici
- Indagini biochimiche annuali:
- PRL, IGF-1 (dall’età di 5 anni);
- Calcemia totale (corretta per l'albumina) e/o calcio ionizzato (dall’età di 8 anni);
- PTH intatto, plasmatico, a digiuno (dagli 8 anni);
- Gastrinemia a digiuno e/o stimolata, se un individuo ha importanti sintomi di reflusso, crisi diarroiche o franca evidenza di sindrome di Zollinger-Ellison (dall’età di 20 anni);
- Glicemia, insulinemia a digiuno, pro-insulina (dall’età di 5 anni)
- Da prendere in considerazione:
- fra i test biochimici: dosaggio del polipeptide pancreatico
- fra i test d’immagine: immagine a livello addominale e RM cranio-sella.
- Test clinici di follow-up per tutta la vita a 3-5 anni d’intervallo (2-4).
È importante che i soggetti affetti da MEN 1 e i loro familiari a rischio siano seguiti presso centri clinici che oltre ad avere un'evidente esperienza specifica per questa patologia, abbiamo in atto anche specifici percorsi diagnostico-clinici, multidisciplinari, per tali soggetti, che siano capaci di prevedere una tempistica ideale di follow-up che si adatti a ciascun paziente MEN1, in base al proprio quadro clinico.
Bibliografia
- Geerdink EA, Van der Luijt RB, Lips CJ. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol 2003, 149: 577–82.
- Falchetti A, Marini F, Luzi E, et al. Multiple endocrine neoplasia type 1 (MEN1): not only inherited endocrine tumors. Genet Med 2009, 11: 825-35.
- Falchetti A. Genetic screening for multiple endocrine neoplasia syndrome type 1 (MEN-1): when and how. F1000 Med Rep 2010, Feb 24: 2.
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001, 86: 5658–71.
- Fendrich V, Langer P, Waldmann J, et al. Management of sporadic and multiple endocrine neoplasia type 1 gastrinomas. Br J Surg 2007, 94: 1331–41.
- Gibril F, Chen YJ, Schrump DS, et al. Prospective study of thymic carcinoids in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2003, 88: 1066-81.
- Sachithanandan N, Harle RA, Burgess JR. Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer 2005, 103: 509–15.
- Bassett JH, Forbes SA, Pannett AA, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 1998, 62: 232–44.
Epidemiologia della MEN-2
Erica Solaroli
UOSD di Endocrinologia, Ospedale Maggiore-Azienda USL di Bologna
Le neoplasie endocrine multiple di tipo 2 (MEN-2) sono ereditate con carattere autosomico dominante e determinate da una mutazione attivante del pro-oncogene RET (1). La prevalenza stimata nella popolazione generale è di 2.5 per 100.000.
La MEN 2 si manifesta in tre varianti cliniche con diversa penetranza di carcinoma midollare della tiroide, feocromocitoma e iperparatiroidismo primitivo (2).
- La MEN-2A copre oltre il 60% dei casi, ed è caratterizzata dalla presenza di carcinoma midollare della tiroide (MTC), feocromocitoma e tumori multipli delle paratiroidi (3). Virtualmente tutti i pazienti con MEN-2A sviluppano MTC, il 40-50% feocromocitoma, il 10-30% iperparatiroidismo. Il MTC è in genere la prima manifestazione clinica, che nella maggior parte dei casi insorge tra i 5 e i 25 anni. Esistono varianti rare, in cui la MEN-2A si associa alla presenza di lichen amiloidosico cutaneo o di malattia di Hirschsprung.
- La MEN-2B interessa il 5-10% dei casi ed è caratterizzata da associazione di MTC, feocromocitoma, neurinomi di labbra e lingua, ganglioneuromi dell’intestino, habitus marfanoide, deformazioni scheletriche e lassità dei legamenti. La MEN-2B non esprime iperparatiroidismo primitivo. Nella MEN-2B il MTC si manifesta nei primi anni di vita (a volte nei primi mesi) e in forma più aggressiva.
- Il carcinoma midollare familiare (FMTC) rappresenta circa il 30% dei casi di MEN-2 ed è caratterizzato da una alta prevalenza di MTC nella stessa famiglia (4 o più casi), con età di comparsa più tardiva (3, 4, 5). Oggi l’FMTC è considerato una variante fenotipica di MEN-2A, con bassissima penetranza di feocromocitoma e iperparatiroidismo primitivo. Una variante rara di FMTC comprende l’associazione con malattia di Hirschsprung.
I dati relativi a 500 famiglie con MEN-2 presenti in Europa fanno rilevare una diversa frequenza delle mutazioni (tabella) (6).
| Frequenza delle mutazioni RET | |
| Codone | % |
| 634 | 34.2 |
| 804 | 17.2 |
| 918 | 10.4 |
| 790 | 7.6 |
| 891 | 7.2 |
| 620 | 7 |
| 618 | 6.4 |
| 791 | 3.2 |
| 768 | 2.6 |
| 609-611 | 1.6 |
| 630 | 1 |
Ad oggi sono state descritte in tutto il mondo oltre 1000 famiglie MEN-2 (7).
Bibliografia
- Raue F, Frank-Raue K. Update multiple endocrine neoplasia type 2. Fam Cancer 2010, 9: 449-57.
- Machens A, Niccoli-Sire P, Hoegel J, et al. Early malignant progression of hereditary medullary thyroid cancer. N Engl J Med 2003, 349: 1517-25.
- Frank-Raue K, Rondot S, Schulze E, et al. Change in the spectrum of RET mutations diagnosed between 1994 and 2006. Clin Lab 2007, 53: 273-82.
- Berndt I, Reuter M, Saller B, et al. A new hot spot for mutations in the RET protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 1998, 83: 770-4.
- LiVolsi VA. C cell hyperplasia/neoplasia. J Clin Endocrinol Metab 1997, 82: 39-41.
- Machens A, Lorenz C, Sekulla C, et al. Molecular epidemiology of multiple endocrine neoplasia 2: implications for RET screening in the new millenium. Eur J Endocrinol 2013, 168: 307-14.
- Wells SA, Pacini F, Robinson BG, et al. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: an update. J Clin Endocrinol Metab 2013, Doi:10.1210/jc.2013-1204.
Clinica della MEN-2
Erica Solaroli
UOSD di Endocrinologia, Ospedale Maggiore-Azienda USL di Bologna
Correlazioni fenotipo-genotipo
Da tempo sono note precise associazioni tra fenotipo (età di insorgenza, aggressività del carcinoma midollare, MTC, presenza o assenza di altre neoplasie) e genotipo (tipo di mutazione germinale RET)(1,2).
Circa il 98% delle famiglie con MEN-2A ha una mutazione RET nell’esone 10 o 11; le mutazioni della cisteina nel codone 634 coprono l’86% delle famiglie.
Le mutazioni più frequenti interessano uno dei cinque residui di cisteina nei codoni 609, 611, 618, 620, 634 (esoni 10 e 11) o nei codoni 790, 791, 768 (esone 13) e nel codone 804 (esone 14)(3).
Il 95% dei pazienti con MEN-2B presenta la sostituzione della treonina con la metionina nel codone 918, esone 16. La mutazione nel codone 883, esone 15, è presente nel 2-3% dei casi. Oltre il 50% dei casi di MEN-2B è determinato da una mutazione ex novo dell’oncogene RET.
Il carcinoma midollare familiare (FMTC) è più comunemente associato a mutazioni nei codoni 609, 611, 618, 620 dell’esone 10, nel codone 768 dell’esone 13 e nel codone 804 dell’esone 14 (1).
L’American Thyroid Association (ATA) nel 2009 ha ridefinito la classificazione delle mutazioni RET basata sulla aggressività del MTC, che prevede quattro classi di rischio (4):
- ATA-D: mutazioni a più alto rischio di sviluppo di MTC in età giovanile, di metastasi, di elevata mortalità correlata alla malattia (codoni 883 e 918);
- ATA C: mutazioni correlate con alto rischio di sviluppare MTC, anche se inferiore al precedente (codone 634);
- ATA B: mutazioni a basso rischio per forme aggressive di MTC (codoni 609, 611, 618, 620, 630);
- ATA A: mutazioni con rischio meno elevato di MTC aggressivo (codoni 768, 790, 791, 804, 891).
L’associazione genotipo-fenotipo è importante per stabilire l’età della tiroidectomia e per definire il tipo di screening da effettuare per le neoplasie associate nelle MEN 2-A e MEN-2B (tabella 1).
| Tabella 1 Comportamento clinico e terapia dei pazienti con diverse mutazioni RET (modificata da 3) |
||||
| Codoni | ||||
| 321, 515, 533, 600, 603, 606, 635, 649, 666, 768, 776, 790, 791, 804, 819, 833, 844, 861, 891, 912 | 609, 611, 618, 620, 630, 631 | 634 | 918, 883 | |
| Classe rischio ATA |
A | B | C | D |
| Sottotipo MEN-2 | FMTC | FMTC/MEN-2A | MEN-2A | MEN-2B |
| Aggressività MTC | Moderata | Intermedia | Alta | Molto alta |
| Età insorgenza MTC | Adulta | Dopo i 5 anni | Prima dei 5 anni | Primo anno |
| Tabella 2 Frequenza dei segni e dei sintomi dei pazienti con feocromocitoma |
|
| Segni e sintomi | Frequenza |
| Cefalea | 60-80% |
| Tachicardia/palpitazioni | 50-70% |
| Sudorazione | 40-60% |
| Ansia | 20-40% |
| Ipertensione severa | 50-60% |
| Ipertensione parossistica | 40-60% |
| Pallore | 35-45% |
| Nausea | 20-25% |
| Perdita di peso | 20-40% |
| Ipotensione ortostatica | 10-20% |
| Ridotta tolleranza glucidica/diabete | 40-50% |
| Flushing | 10-20% |
| Dispnea | 10-20% |
| Vertigini | 10-20% |
Il MTC è stato riscontrato:
- nei bambini con MEN-2B entro il secondo mese di vita;
- nella MEN-2A al decimo mese di vita in presenza di mutazione del codone 634;
- nella MEN-2A o FMTC dipende da quali sono i codoni mutati:
- codone 630: entro il 12° mese;
- codoni 609, 611, 618, 620: tra i 4 e i 7 anni;
- codoni 533, 768, 790, 791, 804 e 891: tra i 9 e i 21 anni.
Le metastasi linfonodali da MTC compaiono in epoca diversa in relazione ai diversi codoni mutati:
- codone 918 (MEN-2B): prima dei 2 anni;
- codone 634: non prima dei 5 anni;
- codone 630: non prima dei 15 anni;
- nelle restanti mutazioni: dopo i 20 anni.
Nella casistica retrospettiva multicentrica del Gruppo di studio francese dei tumori endocrini (5) inerente 170 pazienti con MEN-2 o FMTC, operati di tiroidectomia totale prima dei 21 anni, i pazienti più giovani con MTC (> 10 mm) o metastasi linfonodali avevano rispettivamente:
- quelli con mutazione RET di classe ATA B: 14 e 16 anni;
- quelli in classe ATA C: 13 e 11 anni;
- con classe ATA D: 6 e 2 anni.
Il feocromocitoma è presente:
- in circa il 50% dei pazienti con MEN-2A e MEN-2B, rispettivamente portatori di mutazione nei codoni 634 e 918;
- nel 17% dei pazienti MEN-2A con mutazione nell’esone 10, codoni 609, 611, 618, 620;
- raro in associazione a mutazione nell’esone 13 (codone 791) e 14 (codone 804).
Il feocromocitoma può manifestarsi a età diverse, a seconda dei codoni mutati:
- codone 918 e 634: entro i 12 anni;
- codoni 609, 611, 618 e 620: tra i 19 e 30 anni;
- codoni 768, 790, 791, 804 e 891: tra i 28e i 59 anni.
Tutti i casi di MEN-2A associati a malattia di Hirschsprung presentano mutazioni nell’esone 10, codoni 609, 611, 618, 620.
L’associazione MEN-2A e lichen amiloidosico cutaneo è legata a mutazione del codone 634, esone 11 (6,7).
L’iperparatiroidismo si riscontra in età diverse, a seconda dei codoni mutati (8):
- codone 634: età infantile;
- codoni 609, 611, 618 e 620: tra i 34 e i 41 anni;
- codoni 533, 768, 790, 791, 804 e 891: tra i 38 e i 54 anni.
Clinica
A differenza della forma sporadica di MTC, che si presenta quasi sempre come lesione unica, l’MTC ereditario è multicentrico, si sviluppa nel terzo medio e superiore dei lobi tiroidei e si associa a iperplasia delle cellule C, che ne rappresenta la condizione pre-neoplastica.
Il periodo necessario alla progressione della neoplasia, da tumore ancora confinato alla tiroide, all’interessamento linfonodale, e successivamente alla metastatizzazione a distanza, è variabile e rispecchia il comportamento biologico del tumore.
Il 30% dei pazienti con carcinoma di dimensioni < 1 cm presenta metastasi ai linfonodi del compartimento centrale, latero-cervicali, mediastinici.
Metastasi a distanza interessano prevalentemente polmone, fegato e osso.
Il marcatore neoplastico è la calcitonina, elevata in condizioni basali o stimolata (attualmente è utilizzabile solo il test al calcio gluconato, non essendo più disponibile la pentagastrina).
I livelli basali di calcitonina correlano con le dimensioni della neoplasia (9-12).
Quando si valuta la calcitonina, basale e stimolata, nei portatori di mutazione si deve sempre tener presente che i valori sono maggiori nell’uomo rispetto alla donna (correlati alla maggiore rappresentazione di cellule parafollicolari C nell’uomo), e nei bambini rispetto agli adulti, specie nei bambini di età < 3 anni.
Altro eccellente marcatore neoplastico è la glicoproteina CEA.
Le manifestazioni cliniche del feocromocitoma sono le stesse delle forme sporadiche (tabella 2)(13) e sono correlate alla sintesi, al rilascio di catecolamine (noradrenalina, adrenalina, dopamina) e alla capacità di stimolazione del recettore adrenergico. Il feocromocitoma è un tumore altamente vascolarizzato e spesso presenta aree necrotico/emorragiche, che possono correlare con una minore espressività clinica; una estesa emorragia intra-tumorale può causare una crisi ipertensiva e successiva scomparsa dei sintomi.
Nelle MEN-2A e MEN-2B il feocromocitoma è pressochè sempre benigno, a localizzazione surrenalica, e nel 60% circa dei pazienti che lo esprimono è multicentrico e bilaterale, ha una più alta concentrazione intra-tumorale di catecolamine e una minore concentrazione plasmatica in rapporto alle dimensioni della neoplasia (13).
Questa forma ereditaria di feocromocitoma è asintomatica in percentuale variabile dal 30 al 50%, data la diagnosi più precoce conseguente allo screening.
La diagnosi viene posta dopo quella di MTC in circa il 50% dei pazienti con MEN-2A o MEN-2B, è sincrona in circa il 40% e solo nel 10% la precede.
Nel sospetto di feocromocitoma, o nello screening pre-operatorio (tiroidectomia terapeutica o profilattica, o chirurgia per altre patologie nei pazienti con MEN-2), la valutazione biochimica si basa sul dosaggio di metanefrine plasmatiche e/o metanefrine frazionate urinarie (in questa forma ereditaria sono aumentate metanefrina e normetanefrina).
L’iperparatiroidismo primitivo è determinato da iperplasia/adenoma delle cellule principali di più paratiroidi, spesso è silente e raramente rappresenta la prima manifestazione della sindrome. La diagnosi viene fatta attraverso il rilievo di elevati valori di PTH e calcemia, solitamente dopo la terza decade di vita (14,15).
Il lichen amiloidosico cutaneo, presenta in circa il 10% delle famiglie MEN-2A, è una lesione cutanea pruriginosa, localizzata in sede inter-scapolare (tra T2 e T6), spesso iperpigmentata, che può manifestarsi in giovane età e può precedere la diagnosi di MTC.
La malattia di Hirschsprung è determinata dall’assenza dei gangli dei plessi mio-enterici di Auerbach e sotto-mucoso di Meissner di parte dell’intestino, con conseguenti anomalie della persistalsi e frequente occlusione intestinale; è presente in circa il 7% dei pazienti con MEN-2A o FMTC.
La MEN-2B è caratterizzata dalla precoce comparsa di forme aggressive di carcinoma midollare; il feocromocitoma compare nel 50% dei casi e in circa la metà dei casi è multiplo e spesso bilaterale. Non esprime iperparatiroidismo primitivo.
Nelle forme ex novo la rapida identificazione dei sintomi è indispensabile per una diagnosi precoce. Nei primi mesi di vita compaiono neurinomi mucosi sulla superfice anteriore di lingua, palato, faringe, labbra, palpebre (figura). Circa il 40% dei pazienti presenta una ganglio-neuromatosi diffusa del tratto gastro-intestinale con distensione addominale, megacolon, diarrea alternata a stipsi. Circa l’80% dei pazienti ha habitus marfanoide e anomalie scheletriche, come lordosi, cifosi, cifoscoliosi, lassità dei legamenti, piede equino-varo, deformità del petto (scavato o carenato)(16-18).
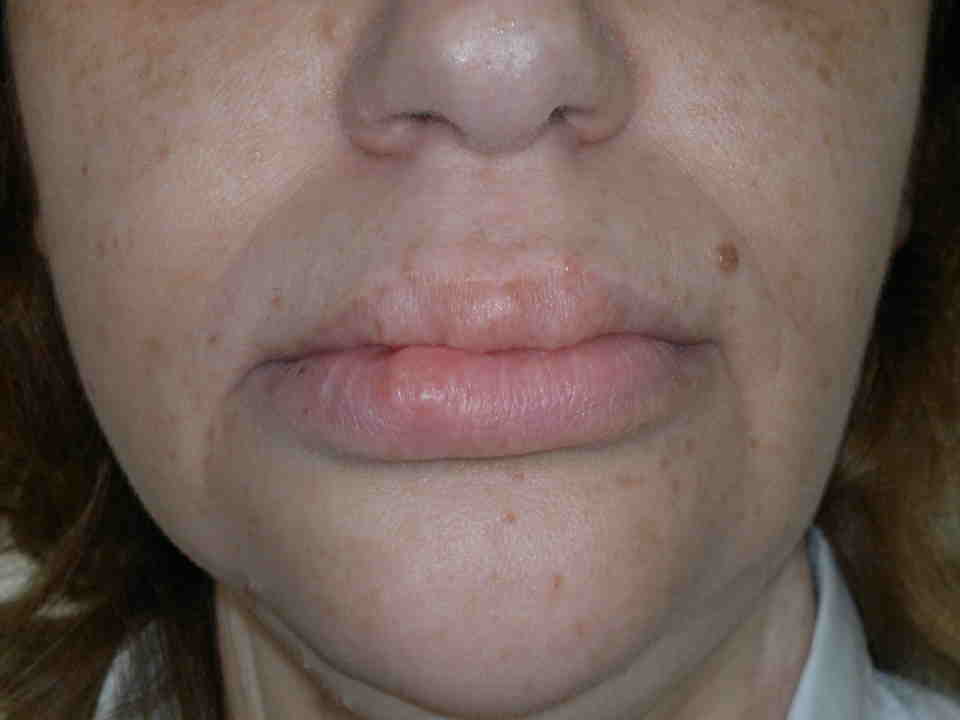
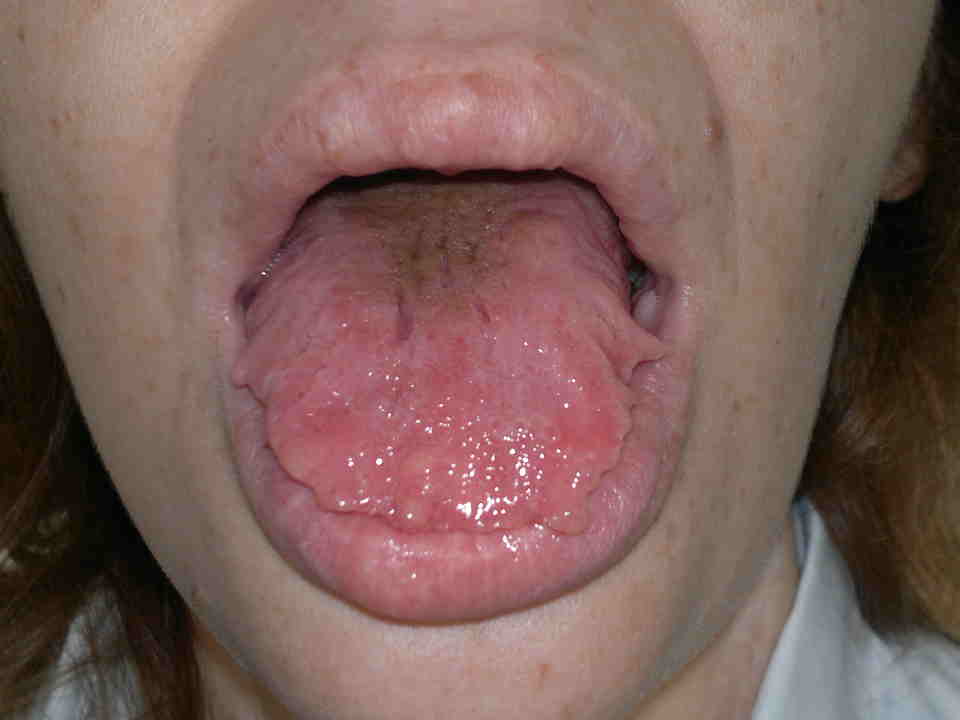
Neurinomi mucosi delle labbra (a sinistra) e della lingua (a destra) in MEN-2B.
Nei soggetti con carcinoma midollare familiare, il carcinoma midollare è la sola manifestazione clinica. La definizione più rigida prevede la trasmissione di carcinoma midollare in più generazioni, in cui nessun membro familiare presenti feocromocitoma o iperparatiroidismo. Una definizione meno rigida prevede la presenza di carcinoma midollare in quattro membri della famiglia affetta. Le controversie sono relative a una prematura categorizzazione di una famiglia con rischio di non fare lo screening, e quindi diagnosi precoce, di feocromocitoma.
La penetranza per il carcinoma midollare è più bassa e l’età di insorgenza è ritardata rispetto a quella della MEN-2A e MEN-2B (tabella 3) (18).
| Tabella 3 Caratteristiche cliniche di MEN-2A, MEN-2B, FMTC |
|||
| Clinica | Carcinoma midollare | Ereditarietà | Patologie associate |
| MEN-2A | Multifocale bilaterale | Autosomica dominante | Feocromocitoma (~50%) Iperparatiroidismo (10-30%) Lichen amiloidosico cutaneo (raro) Malattia di Hirschsprung (raro) |
| MEN-2B | Multifocale bilaterale | Autosomica dominante | Feocromocitoma (~50%) Multipli neurinomi mucosi (>95%) Habitus marfanoide (~80%) |
| FMTC | Multifocale bilaterale | Autosomica dominante | Variante con malattia di Hirschsprung (rara) |
Bibliografia
- Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996, 276: 1575-9.
- Frank-Raue K, Hoppner W, Frilling A, et al. Mutations of the RET protooncogene in German multiple endocrine neoplasia families: relation between genotype and phenotype. German Medullary Thyroid Carcinoma Study Group. J Clin Endocrinol Metab 1996, 81: 1780-3.
- Raue F, Frank-Raue K. Genotype-phenotype correlation in multiple endocrine neoplasia type 2. Clinics 2012, 67 (S1): 69-75.
- Kloos RT, Eng C et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19: 565-612.
- Rohmer V, Vidal-Trecan G, Bourdelot A, et al. Prognostic factors of disease-free survival after thyroidectomy in 170 patents with a RET germline mutation: a multicentre study of the Groupe Francais d’Etude des Tumeurs Endocrines. J Clin Endocrinol Metab 2011, 96: E509-18.
- Schuffenecker I, Virally-Monod M, Brohet R, et al. Risk and penetrance of primary hyperparathyroidism in multiple endocrine neoplasia type 2A families with mutations at codon 634 of the RET proto-oncogene. Groupe D’etude des Tumeurs a Calcitonine. J Clin Endocrinol Metab 1998, 83: 487-91.
- Kouvaraki MA, Shapiro SE, Perrier ND, et al. RET proto-oncogene: a review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 2005, 15: 531-44.
- Machens A, Dralle H. Multiple endocrine neoplasia type 2: achievements and current challenges. Clinics 2012, 67 (S1): 113-8.
- Romei C, Cosci B, Renzini G, et al. RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin Endocrinol (Oxf) 2011, 74: 241-7.
- Costante G, Meringolo D, Durante C, et al. Predictive value of serum calcitonin levels for preoperative diagnosis of medullary thyroid carcinoma in a cohort of 5817 consecutive patients with thyroid nodules. J Clin Endocrinol Metab 2007, 92: 450-5.
- Doyle P, Duren C, Nerlich K, et al. Potency and tolerance of calcitonin stimulation with high-dose calcium versus pentagastrin in normal adults. J Clin Endocrinol Metab 2009, 94: 2970-4.
- Cohen R, Campos JM, Salaun C, et al. Preoperative calcitonin levels are predictive of tumor size and postoperative calcitonin normalization in medullary thyroid carcinoma. Groupe d’Etudes des Tumeurs a Calcitonine (GETC). J Clin Endocrinol Metab 2000, 85: 919-22.
- Mannelli M, Lenders JWM, Pacak K, et al. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab 2012, 26: 507-15.
- Kraimps JL, Denizot A, Carnaille B, et al. Primary hyperparathyroidism in multiple endocrine neoplasia type IIa: retrospective French multicentric study. Groupe d’Etude des Tumeurs a Calcitonine, French Association of Endocrine Surgeons. World J Surg 1996, 20: 808-12; discussion 12-3.
- Raue F, Kraimps JL, Dralle H, et al. Primary hyperparathyroidism in multiple endocrine neoplasia type 2A. J Intern Med 1995, 238: 369-73.
- Wray CJ, Rich TA, Waguespack SG, et al. Failure to recognize multiple endocrine neoplasia 2B: more common than we think? Ann Surg Oncol 2008,15: 293–301.
- Moline J, Eng C. Multiple endocrine neoplasia type 2: An overview. Genet Med 2011, 13: 755-64.
- Wu LS, et al. Medullary thyroid cancer: an update of new guidelines and recent developments. Curr Opin Oncol 2011, 23: 22–7.
Dal sospetto alla diagnosi di MEN-2
Rossella Elisei & Cristina Romei
Endocrinologia, Università di Pisa
Per poter identificare una forma familiare, il paziente affetto da carcinoma midollare della tiroide (MTC) dovrebbe essere sottoposto ad un attento esame della storia medica familiare. Qualora nell’ambito di una stessa famiglia fossero presenti più soggetti (almeno in 4 generazioni) affetti da MTC o altri tumori quali feocromocitoma (PHEO) e/o iperplasia/adenomatosi delle ghiandole paratiroidee (PHPT), il sospetto di una forma familiare diviene molto alto e il test genetico sicuramente appropriato (1,2). Ad eccezione di alcune rare mutazioni che non sembrano avere una reale attività trasformante (3), la presenza di una mutazione germinale di RET oggi permette di fare la diagnosi di MEN-2 anche se la storia familiare è apparentemente negativa.
Dopo aver effettuato la diagnosi genetica di MEN-2, il paziente deve essere indagato per la presenza delle diverse patologie endocrine. Dal momento che mutazioni diverse possono essere associate a fenotipi diversi in cui possono essere coinvolte neoplasie diverse, la diagnosi clinica per la presenza di PHEO e/o PHPT viene suggerita dal tipo di mutazione (2, 4, 5). In ogni modo le procedure per la diagnosi di MTC, PHEO e PHPT in pazienti affetti da MEN-2 sono quelle utilizzate per i pazienti affetti dalle corrispondenti forme sporadiche.
MTC
La CT serica è un marcatore molto sensibile e specifico per l’MTC, anche se altre condizioni patologiche possono talvolta essere accompagnate da un aumento dei livelli di CT (6,7). Il test di stimolo, sia con pentagastrina che con calcio, è dirimente per discriminare una produzione di CT causata dall’MTC dalla produzione di CT dovuta ad altre condizioni (6,7). È stato largamente dimostrato che importanti fattori prognostici negativi per l’MTC sono una fase avanzata del tumore al momento della diagnosi e, in particolare, la presenza di metastasi linfonodali e/o a distanza. In considerazione di ciò, si può affermare che solo una diagnosi precoce seguita da un trattamento chirurgico completo rappresentano l’unica opportunità di guarigione per questi pazienti (8,9).
L’esame citologico del nodulo tumorale ed il dosaggio della CT basale e stimolata sono gli strumenti diagnostici più importanti per la diagnosi di MTC (10). Valori serici basali di CT superiori al range di normalità dovrebbero essere considerati come positivi e suggestivi, anche se non necessariamente diagnostici, della presenza di MTC. A questo proposito è bene ricordare che ciascun centro dovrebbe avere il suo range di normalità, calcolato misurando la CT nel siero di soggetti con assenza di malattie della tiroide. Il dosaggio della CT, dosaggio che è consigliabile eseguire solo in presenza di noduli tiroidei, nel caso dell’MTC ha una attendibilità diagnostica maggiore di quella dell’esame citologico su agoaspirato tiroideo (10), il quale non sempre risulta dirimente. In questi casi, a completamento dell’iter diagnostico di questo tumore, è comunque opportuno eseguire la misurazione della CT nel liquido di lavaggio dell’ago utilizzato per il prelievo citologico (11). In ogni caso, qualora sia l’esame citologico dell’agoaspirato che la misurazione della CT sul liquido di lavaggio dell’ago usato per la citologia dovessero non essere dirimenti, prima di sottoporre il paziente ad intervento chirurgico è opportuno eseguire una stimolazione della CT mediante somministrazione ev di pentagastrina o calcio: un incremento della CT di almeno 4 volte il valore basale, più che il valore del picco di stimolazione per sè, sarà fortemente suggestivo di MTC (12).
Dal momento che le sindromi MEN-2 sono costituite dall’associazione dell’MTC con altre patologie endocrine, il percorso diagnostico deve essere completato anche con indagini cliniche volte ad identificare sia il PHEO che il PHPT, soprattutto nei casi RET-positivi per quelle mutazioni note per predisporre allo sviluppo di queste patologie.
PHEO
Il PHEO è presente in circa il 40-50% dei pazienti affetti da MEN-2, sia di tipo A che di tipo B (13). Poiché il PHEO può essere asintomatico nei pazienti con MEN-2, può essere difficile escluderlo con certezza. Lo screening per il PHEO è costituito oggi dalla misurazione delle metanefrine, o laddove questa valutazione non sia ancora disponibile, delle catecolamine plasmatiche e urinarie. Quando i livelli di metanefrine e/o catecolamine risultino elevati, è suggerito uno studio di imaging (TC o RMN). Tutti i pazienti con evidenza di eccesso di catecolamine dovrebbero inoltre essere trattati farmacologicamente prima di qualsiasi trattamento chirurgico, per il rischio di sviluppare gravi crisi ipertensive durante lo stress chirurgico se non preparati con farmaci alfa- e beta-bloccanti. Anche i pazienti con dimostrata presenza di lesioni surrenaliche ma senza evidenza biochimica della malattia, dovrebbero essere sottoposti a blocco adrenergico.
Quando il PHEO è presente simultaneamente alle altre patologie, deve essere rimosso per primo, perché, anche se asintomatico, aumenta notevolmente il rischio dell'intervento chirurgico per la rimozione dell’MTC e del PHPT (14,15).
PHPT
Circa il 20-30% dei pazienti affetti da MEN-2A presenta iperplasia o adenomatosi multipla delle paratiroidi. La prevalenza maggiore si ha nei casi di MEN-2A con mutazioni di RET che interessano il codone 634 dell’esone 11 (5) e in particolare con la mutazione C634A. La manifestazione clinica più comune della patologia delle paratiroidi è l’iperparatiroidismo, che porta a un aumento dei livelli ematici di calcio a seguito di una maggiore rimozione del calcio dall’osso e conseguente osteoporosi. Spesso, e soprattutto all’inizio della patologia, la presenza di PHPT decorre asintomatica e pertanto lo studio di questa patologia deve comunque essere effettuato nei pazienti RET-positivi.
Le procedure diagnostiche per l’iperparatiroidismo in pazienti MEN-2 sono le stesse usate anche per i pazienti affetti da iperparatiroidismo di tipo sporadico. La prima indagine sarà la misurazione del paratormone (PTH) sierico e della calcemia. Qualora si dovesse verificare il caso di PTH elevato con calcemia ancora normale, occorre escludere la presenza di un iperparatiroidismo secondario, peraltro oggi frequente a causa della frequente ipovitaminosi D. Se al termine di tutte le valutazioni viene posta la diagnosi di iperparatiroidismo primario, occorrerà valutare i livelli di calciuria e l’eventuale presenza di calcolosi renale e/o di osteoporosi. È sempre indicata un’ecografia del collo, anche se talvolta può risultare negativa.
Bibliografia
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001, 86: 5658-71.
- Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996, 276: 1575-9.
- Cosci B, Vivaldi A, Romei C, et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr Relat Cancer 2011, 18: 603-12.
- Figlioli G, Landi S, Romei C, et al. Medullary thyroid carcinoma (MTC) and RET proto-oncogene: mutation spectrum in the familial cases and a meta-analysis of studies on the sporadic form. Mutat Res 2013, 752: 36-44.
- Raue F, Frank-Raue K. Genotype-phenotype relationship in multiple endocrine neoplasia type 2. Implications for clinical management. Hormones (Athens) 2009, 8: 23-8.
- Elisei R. Routine serum calcitonin measurement in the evaluation of thyroid nodules. Best Pract Res Clin Endocrinol Metab 2008, 22: 941-53.
- Elisei R, Romei C. Calcitonin estimation in patients with nodular goiter and its significance for early detection of MTC: european comments to the guidelines of the American Thyroid Association. Thyroid Res 2013, 6 Suppl 1: S2.
- Pelizzo MR, Boschin IM, Bernante P, et al. Natural history, diagnosis, treatment and outcome of medullary thyroid cancer: 37 years experience on 157 patients. Eur J Surg Oncol 2007, 33: 493-7.
- Gharib H, McConahey WM, Tiegs RD, et al. Medullary thyroid carcinoma: clinicopathologic features and long-term follow-up of 65 patients treated during 1946 through 1970. Mayo Clin Proc 1992, 67: 934-40.
- Bugalho MJ, Santos JR, Sobrinho L. Preoperative diagnosis of medullary thyroid carcinoma: fine needle aspiration cytology as compared with serum calcitonin measurement. J Surg Oncol 2005, 91: 56-60.
- Trimboli P, Cremonini N, Ceriani L, et al. Calcitonin measurement in aspiration needle washout fluids has higher sensitivity than cytology in detecting medullary thyroid cancer: a retrospective multicenter study. Clin Endocrinol 2013, DOI: 10.1111/cen.12234.
- Pacini F, Fontanelli M, Fugazzola L, et al. Routine measurement of serum calcitonin in nodular thyroid diseases allows the preoperative diagnosis of unsuspected sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1994, 78: 826-9.
- Howe JR, Norton JA, Wells SA Jr. Prevalence of pheochromocytoma and hyperparathyroidism in multiple endocrine neoplasia type 2A: results of long-term follow-up. Surgery 1993, 114: 1070-7.
- Walz MK, Alesina PF. Single access retroperitoneoscopic adrenalectomy (SARA)--one step beyond in endocrine surgery. Langenbeck Arch Surgery 2009, 394: 447-50.
- Romei C, Pardi E, Cetani F, Elisei R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J Oncol 2012, 2012: 705036.
Screening biochimico e radiologico della MEN-2: quando e come
Nadia Cremonini
UOSD di Endocrinologia – Ospedale Maggiore – Azienda USL di Bologna
Nei portatori di mutazione germinale di RET, lo screening per le patologie che possono sviluppare deve essere guidato dalla mutazione presente, data la correlazione esistente tra genotipo e fenotipo nelle MEN-2.
Carcinoma midollare della tiroide (MTC)
Quando si effettua il dosaggio di calcitonina (Ct) in bambini di età < 3 anni, e in particolare < 6 mesi, l’interpretazione dei valori risulta difficile, in quanto fisiologicamente sono più elevati di quelli dell’adulto.
Nei bambini con MEN-2B (livello di rischio ATA D) che vengono inviati alla tiroidectomia profilattica prima dei sei mesi, la determinazione pre-operatoria di Ct è opzionale, in quanto il suo ruolo non è ben definito, mentre è necessaria nei bambini di età > di 6 mesi non ancora operati, e per questi si rende necessaria anche la US del collo, per escludere metastasi linfonodali e quindi modificare il tipo di intervento (1).
Anche per i bambini con mutazioni di RET dei livelli di rischio ATA A (515, 531, 600, 603, 777, 912, a espressione clinica FMTC, e 533, 649, 666, 768, 790, 791, 804, 891, con espressione clinica MEN-2A), B (codoni 609, 611, 618, 620, 630, 631, 633, espressione clinica MEN-2A) e C (c634, MEN-2A), inviati alla tiroidectomia profilattica tra 3 e 5 anni, il dosaggio di Ct pre-intervento è considerato opzionale (1), così come la US tiroidea.
Dopo i 5 anni sono raccomandate entrambe le valutazioni, per rilevare metastasi linfonodali, con conseguente pianificazione di intervento più esteso.
Nel caso l’intervento di tiroidectomia profilattica venga rimandato a età > 5 anni (condizioni necessarie: normali livelli di Ct basale (bCt) e dopo stimolo (sCt), normale US collo, bassa aggressività di MTC nei familiari affetti, preferenza dei genitori), il controllo di Ct basale e dopo stimolo e di US collo vanno effettuati annualmente. Se rilevati linfonodi patologici, è indicato effettuare TC o RM di collo e mediastino.
Secondo le Linee Guida ATA, il ruolo dei test di stimolo annuali (ora è possibile effettuare solo il test al calcio gluconato) è meno definito, e viene solo considerata la possibilità di effettuarli (1). È più recente la segnalazione in letteratura dell’importanza di bCt (2) o di bCt basale e sCt (3) per stabilire il momento più idoneo per l’intervento di tiroidectomia nei bambini portatori di mutazioni RET di classe di rischio ATA A, B, C (intervento indicato nel momento in cui la bCt o sCt diventa positiva): in queste casistiche l’outcome è risultato analogo a quello osservato con la tradizionale tiroidectomia profilattica basata unicamente su genotipo ed età. Se si può concordare con questo atteggiamento per i gene carriers di classe A o B, si ritiene necessario disporre di maggiori dati per la classe di rischio C.
L’ecografia del collo è importante soprattutto in fase pre-operatoria, o qualora si rilevino valori elevati di bCt, per valutare la presenza di metastasi linfonodali e definire l’estensione dell’intervento chirurgico, mentre non ha valore come indagine predittiva di presenza o assenza di MTC nei bambini gene carriers di mutazione RET per MEN-2A. Morris et al. in 35 bambini MEN-2A sottoposti a tiroidectomia in età compresa tra 3 e 13.8 anni (mediana 6.39) hanno riscontrato una sensibilità di US nel predire la presenza di MTC del 13% [IC95% 2-40%] e specificità del 95% [IC95% 75-100%] per noduli ≥ 5 mm, mentre rimuovendo tale cut-off dimensionale la sensibilità aumentava al 27% [IC95% 8-55%], accanto però a diminuzione della specificità al 70% [IC95% 46-88%] (4).
Feocromocitoma (FCC)
La valutazione biochimica va effettuata annualmente mediante determinazione di metanefrine plasmatiche frazionate, o se non disponibile tale metodica, con metanefrine urinarie (1,7).
Nella MEN-2 il FCC si esprime raramente nell’infanzia e le Linee Guida ATA hanno indicato l’età di inizio dello screening sulla base dell’età di esordio della patologia correlata alle diverse mutazioni di RET (1):
- 8 anni, nei gene carriers di mutazioni RET associate a MEN-2B e delle mutazioni nei codoni 630 e 634 (MEN-2A);
- 20 anni, per i gene carriers di altre mutazioni RET correlate a MEN-2A;
- 20 anni per i gene carriers di mutazioni associate solo a FMTC.
Viene raccomandato uno screening annuale per FCC per i pazienti con MEN-2A, in particolare per i portatori di mutazione c634, e per i pazienti con MEN-2B (1), dato l’elevato rischio di forme bilaterali (fino al 60–65%) nei soggetti che manifestano FCC, e in tal caso lo sviluppo di FCC controlaterale avviene generalmente entro 10 anni (5).
Le linee guida ATA indicano uno screening “periodico”, non ulteriormente specificato, per i pazienti FMTC: in questi pazienti si ritiene ragionevole indicare uno screening biochimico biennale a partire dai 30 anni.
La valutazione di una casistica tedesca di 474 gene carriers ha rilevato un’età media alla prima surrenectomia per FCC di 25.5 anni (range 19–31 anni) per i pazienti di classe ATA D, 34.7 anni (range 14–67) per la classe C, 40.5 anni (range 22–57) per la classe B e 56.5 anni (range 39-72) per la classe A (6).
In base alla maggiore penetranza di FCC nelle varie classi di rischio ATA e al picco di esordio, questi Autori hanno considerato un comportamento più flessibile per lo screening di FCC (6):
- classe D: iniziare a 11 anni, biennale sino ai 20 anni, indi annuale sino ai 40; viene posto il dubbio se si renda necessario dopo tale età;
- classe C: iniziare a 11 anni, biennale fino ai 30 anni e dai 41 ai 70, annuale tra i 31 e i 40 anni;
- classe B: iniziare a 16 anni, biennale fino ai 30 anni e dai 51 ai 60, e annuale tra i 31 e i 50 anni;
- classe A: iniziare a 26 anni, indi biennale sino ai 70 anni o oltre.
Nonostante sia noto il picco di esordio di FCC nei pazienti con MEN-2 in alcune decadi di vita, sono necessarie casistiche più ampie per poter indicare uno screening meno frequente, es. biennale, al di fuori di tali periodi, per le classi di rischio D, C, B.
Le donne con mutazione RET associata a MEN-2 devono essere sottoposte a screening per FCC prima di iniziare una gravidanza, o all’inizio di una gravidanza non pianificata (1,8).
Una volta posta la diagnosi di FCC, deve essere valutata attentamente la possibilità di malattia bilaterale mediante le indagini di imaging.
Lo screening radiologico per FCC non è raccomandato in assenza di clinica e/o dati biochimici sospetti.
Iperparatiroidismo primario (PHP)
Lo screening biochimico prevede la determinazione annuale di calcio corretto per albumina o calcio ionizzato (con o senza PTH).
Il PHP è molto raro nei bambini MEN-2A, e in tal caso associato a mutazione nel codone 634; le linee guida ATA indicano di iniziare lo screening (1):
- a 8 anni nei gene carriers di mutazioni nei codoni 630 e 634 (MEN-2A);
- a 20 anni per i gene carriers di altre mutazioni RET correlate a MEN-2A;
- a 20 anni per i gene carriers di mutazioni associate solo a FMTC.
Lo screening non è necessario per i pazienti MEN-2B, in quanto non sviluppano patologia delle paratiroidi.
Lo screening è annuale nei pazienti MEN-2A; per i pazienti FMTC le linee guida ATA indicano uno screening “periodico”, non meglio definito: in questi pazienti si ritiene ragionevole indicare uno screening biennale.
Conclusioni
A prescindere dalla mutazione RET e dall’età, nei gene carriers per i quali viene diagnosticato il MTC, lo screening per FCC e PHP deve sempre essere effettuato prima dell’intervento per MTC.
Se una famiglia presenta caratteri clinici di MEN-2A o MEN-2B o FMTC, ma in assenza di rilievo di mutazione di RET, i componenti devono essere sottoposti a screening biochimico per MTC, FCC, PHP e a US tiroide, con frequenza di 1-3 anni in base all’età di presentazione delle patologie nel probando e in altri familiari, sino ai 50 anni o ad un’età di 20 anni superiore a quella del componente della famiglia con età più avanzata al momento della diagnosi (1).
Bibliografia
- Kloos RT, Eng C, Evans DB, et al. The American Thyroid Association Guidelines Task Force. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19: 565-612.
- Rohmer V, Vidal-Trecan G, Bourdelot A, et al. Prognostic factors of disease-free survival after thyroidectomy in 170 young patients with a RET germline mutation: a multicenter study of the Groupe Franḉais d’Etude des Tumeurs Endocrines. J Clin Endocrinol Metab 2011, 96: 509-18.
- Elisei R, Romei C, Renzini G, et al. The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab 2012, 97: 426-35.
- Morris LF, Waguespack SG, Edeiken-Monroe BS, et al. Ultrasonography should not guide the timing of thyroidectomy in pediatric patients diagnosed with multiple endocrine neoplasia syndrome 2A through genetic screening. Ann Surg Oncol 2013, 20: 53-9.
- Lairmore TC, Ball DW, Baylin ST, et al. Management of pheochromocytomas in patients with multiple endocrine neoplasia type 2 syndromes. Ann Surg 1993, 217: 595-601.
- Machens A, Lorenz K, Dralle H. Peak incidence of pheochromocytoma and primary hyperparathyroidism in multiple endocrine neoplasia 2: need for age-adjusted biochemical screening. J Clin Endocrinol Metab 2013, 98: 336-45.
- Pacak K, Eisenhofer G, Ahlman H, et al. Pheochromocytoma: recommendations for clinical practice from the First International Symposium. October 2005. Nat Clin Pract Endocrinol Metab 2007, 3: 92-102.
- Elisei R, Alevizaki M, Conte-Devolx B, et al. 2012 European Thyroid Association Guidelines for genetic testing and its clinical consequences in medullary thyroid cancer. Eur Thyroid J 2012, 1: 216-31.
Tiroidectomia profilattica nell'ambito della MEN-2: a chi e quando
Rossella Elisei & Cristina Romei
Endocrinologia, Università di Pisa
Il trattamento terapeutico di scelta nei pazienti affetti da carcinoma midollare della tiroide (MTC) sporadico e familiare è di principio la tiroidectomia totale associata a svuotamento linfonodale del compartimento centrale. La dissezione degli altri compartimenti linfonodali dovrebbe essere invece eseguita solo se coinvolti clinicamente (1). È tuttavia ancora controverso se la dissezione del compartimento centrale debba essere eseguita di routine anche nel caso di giovani pazienti RET Gene Carrier (GC), che non presentano alcuna evidenza clinica e biochimica di malattia (CT basale e dopo stimolo con pentagastrina o calcio, indosabile). Recentemente è stato riportato che non sono mai state ritrovate metastasi linfonodali del comparto centrale nei casi di MTC operati quando i valori di CT erano ancora < 30-40 pg/mL (2). Una situazione analoga è stata osservata da gruppi indipendenti nel caso dei GC e quindi oggi alcune linee guida suggeriscono di evitare la dissezione del comparto centrale specialmente nei GC con CT indosabile o < 30-40 pg/mL (2), dato anche il maggior rischio di lesioni ricorrenziali e/o ipoparatiroidismo post-chirurgico con tale dissezione.
Nella MEN-2, l'età di insorgenza dell’MTC è diversa nelle varie sindromi (3):
- in genere nella prima infanzia per la MEN-2B;
- prevalentemente nella seconda infanzia/adolescenza per la MEN-2A;
- in età adulta per l’FMTC.
Al diverso tempo di latenza corrisponde anche una diversa aggressività della malattia, tanto che bambini affetti da MEN-2B possono presentare malattia metastatica diffusa già durante l’infanzia e, almeno fino agli anni antecedenti lo screening genetico, molti di loro decedevano prima o durante l’adolescenza.
La possibilità di intervenire precocemente è una condizione fondamentale per la guarigione del paziente, poiché la presenza di malattia extra-tiroidea e le metastasi linfonodali sono la causa più frequente di cronicizzazione della malattia e talvolta ulteriore espansione ad organi vitali e conseguente morte nei pazienti con MEN-2A (4). Pertanto, l’obiettivo dello screening genetico è quello di identificare precocemente i GC e intervenire profilatticamente (prima che la malattia si sviluppi) o comunque quando la malattia è ancora intra-tiroidea. Tuttavia, il diverso tempo di latenza che si osserva nello sviluppo dell’MTC, che talvolta è abbastanza lungo, fornisce un’ampia finestra di tempo in cui intervenire, quando le mutazioni vengono identificate prima che il tumore sia manifesto. Recentemente l’American Thyroid Association (ATA) ha pubblicato linee guida per la diagnosi ed il trattamento dell’MTC ed ha classificato le mutazioni di RET in base all’associazione con l’aggressività del tumore (1). Lo scopo del sistema di classificazione è stato quello di offrire raccomandazioni sull'età a cui eseguire la tiroidectomia profilattica. Le mutazioni del gruppo D (i. e. Met918) sono definite a rischio molto alto e sono associate con un’insorgenza precoce della malattia come accade nella MEN-2B. Le mutazioni del gruppo C (i.e. Cys634 e altre) sono definite ad alto rischio e sono più frequentemente legate alla MEN-2A; quelle del gruppo B sono invece a basso rischio e quelle del gruppo A sono definite a rischio molto basso e in entrambi questi gruppi sono collocate la maggior parte delle mutazioni legate al fenotipo FMTC. In generale, l’ATA suggerisce che tutti i soggetti portatori di mutazione di RET, con la sola esclusione delle mutazioni di gruppo A, dovrebbero essere sottoposti a tiroidectomia profilattica (1):
- GC con mutazioni del gruppo D (MEN-2B) dovrebbero essere operati il prima possibile, nel primo anno di vita;
- GC con mutazioni del gruppo C dovrebbero essere sottoposti ad intervento chirurgico entro il quinto anno di vita;
- nei GC con mutazioni di RET appartenenti ai gruppi A e B l’intervento di tiroidectomia può essere eseguito anche dopo i 5 anni di età, fintanto che la patologia non si manifesta con valori di CT positivi.
Partendo però dal presupposto che il MTC, anche di minime dimensioni, è sempre associato a livelli dosabili di CT sierica, la programmazione della tiroidectomia nei GC dovrebbe tenere in considerazione anche il dosaggio della CT, per evitare di sottoporre a intervento chirurgico bambini piccoli nei quali invece la malattia non si svilupperà prima dell’età adulta o comunque dopo il completamento dello sviluppo somatico. Come sopra detto, è stata recentemente riportata una correlazione positiva tra i valori di CT basale e dimensione del tumore. In particolare non sono stati mai riportati tumori di diametro > 0.5 cm con metastasi linfonodali associati a valori di CT < 30-60 pg/mL (2,5). Tali valori di CT variano nei diversi ospedali e ciascun laboratorio dovrebbe avere il suo cut-off di CT in grado di distinguere micro-MTC da tumori più grandi associati con metastasi linfonodali. Tenendo in considerazione quanto detto, si potrebbe ipotizzare di considerare il valore della CT basale nel pianificare l’età a cui eseguire la tiroidectomia, indipendentemente dal tipo di mutazione di RET (6). In ogni caso se la CT basale è al di sotto del range di riferimento del laboratorio, si può evitare la linfoadenectomia del comparto centrale, che rappresenta un rischio elevato per le complicanze chirurgiche, o addirittura si può posticipare l’intervento chirurgico almeno fino al momento in cui il test alla pentagastrina o al calcio non sarà positivo. In quest’ottica il test di stimolo con pentagastrina o calcio rappresenta una procedura diagnostica utile nel pianificare la tiroidectomia senza correre alcun rischio per il paziente.
Bibliografia
- Kloos RT, Eng C, Evans DB, et al. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19: 565-612.
- Elisei R, Romei C, Renzini G, et al. The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab 2012, 97: 426-35.
- Pelizzo MR, Boschin IM, Bernante P, et al. Natural history, diagnosis, treatment and outcome of medullary thyroid cancer: 37 years experience on 157 patients. Eur J Surg Oncol 2007, 33: 493-7.
- Skinner MA, Moley JA, Dilley WG, et al. Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 2005, 353: 1105-13.
- Rohmer V, Vidal-Trecan G, Bourdelot A, et al. Prognostic factors of disease-free survival after thyroidectomy in 170 young patients with a RET germline mutation: a multicenter study of the Groupe Francais d'Etude des Tumeurs Endocrines. J Clin Endocrinol Metab 2011, 96: E509-18.
- Machens A, Dralle H. Multiple endocrine neoplasia type 2 and the RET protooncogene: from bedside to bench to bedside. Mol Cell Endocrinol 2006, 247: 34-40.
Chirurgia per il feocromocitoma nell'ambito della MEN-2
Marco Boniardi
SC di Chirurgia Generale Oncologica e Laparoscopica, Ospedale Niguarda Ca’ Granda – Milano
INDICAZIONI
Il riscontro di un feocromocitoma nell’ambito di una MEN-2 costituisce una precisa indicazione al trattamento chirurgico. Anche nelle forme asintomatiche, che rappresentano circa il 50% dei casi (1), è opportuno procedere all’exeresi della lesione surrenalica come prevenzione di crisi ipertensive, che, nelle forme più gravi, possono esitare in gravi complicanze, quali infarto miocardico, edema polmonare o crisi epilettiche.
Nei rari casi di MEN-2 in cui il feocromocitoma è diagnosticato durante la gravidanza, la paziente deve essere operata nel secondo trimestre: in letteratura vengono riportati più di cento casi trattati con successo, con basse percentuali di morbilità per la madre e per il feto. Nel primo trimestre viene privilegiata la terapia medica, mentre nei feocromocitomi riscontrati nel terzo trimestre è preferibile programmare il parto cesareo con contemporanea exeresi della lesione surrenalica (2).
La preparazione medica all’intervento chirurgico si avvale soprattutto di farmaci alfa-litici (doxazosin, fenossibenzamina), eventualmente associati in una fase successiva a ß-bloccanti (propranololo).
Il trattamento farmacologico con alfa-litici è utile per il controllo dell’ipertensione arteriosa, se presente, ma è soprattutto necessario per la prevenzione dei picchi ipertensivi intra-operatori conseguenti alla manipolazione del tumore.
La necessità di una terapia farmacologica pre-operatoria non è tuttavia universalmente condivisa: alcuni centri di chirurgia endocrina tendono a limitarla alle forme sintomatiche o alle lesioni voluminose.
TERAPIA CHIRURGICA
La terapia chirurgica costituisce attualmente l’unica possibilità di cura radicale del feocromocitoma. Fino agli anni Novanta l’intervento chirurgico di surrenectomia veniva abitualmente condotto con tecnica tradizionale “open”, mediante ampie laparotomie sottocostali, mono o bilaterali, o attraverso lombotomie.
Con l’avvento della chirurgia laparoscopica, l’approccio mini-invasivo si è progressivamente imposto anche nel trattamento delle affezioni benigne del surrene ed è oggi considerato la tecnica di scelta anche per la cura del feocromocitoma (3, 4). Tale approccio presenta indiscutibili vantaggi rispetto alle procedure tradizionali
- percentuale inferiore di complicanze, soprattutto in termini di infezione di ferita e di sviluppo di laparoceli post-operatori;
- miglior controllo del dolore post-operatorio e minor richiesta di farmaci analgesici;
- più rapida ripresa delle funzioni gastro-intestinali;
- riduzione della degenza ospedaliera con più rapido ritorno all’attività lavorativa e alla vita sociale;
- miglior risultato estetico, confrontando le 3-4 piccole ferite della chirurgia endoscopica con le ampie laparotomie sottocostali degli interventi “open”.
La chirurgia tradizionale laparotomica viene oggi riservata ai feocromocitomi di grandi dimensioni, con diametro > 10 cm, per motivi tecnici (difficoltà nella mobilizzazione del tumore e difficile controllo dei peduncoli vascolari) e per il rischio che la lesione possa presentare carattere di malignità. In questo caso, qualora si verificasse un’effrazione del tumore durante le manovre di dissezione, potrebbe essere favorita la disseminazione di cellule neoplastiche (“spillage”) ad opera dei gas (CO2) introdotti in addome per creare la camera di lavoro.
Attualmente l’approccio mini-invasivo al surrene si avvale di due differenti tecniche chirurgiche, che si differenziano sostanzialmente nella via di accesso entrambe laparoscopiche: la via trans-peritoneale e la via retro-peritoneale.
La tecnica trans-peritoneale prevede l’impiego di piccole incisioni cutanee sulla parete addominale, 4 per la surrenectomia destra, 3 per l’asportazione del surrene sinistro, ciascuna della lunghezza di 5–12 mm, utilizzate per l’introduzione, attraverso i trocar, dell’ottica e degli strumenti di lavoro.
La tecnica retro-peritoneale prevede un accesso diretto al surrene, utilizzando tre brevi incisioni cutanee (5–12 mm) in sede lombare. Essa presenta il vantaggio di aggredire direttamente la ghiandola surrenale nello spazio retro-peritoneale, senza necessità di mobilizzare il fegato a destra ed il complesso spleno-pancreatico a sinistra, per accedere alla loggia surrenalica: ciò si traduce in una riduzione dei tempi operatori e in un più rapido decorso post-operatorio (il paziente può assumere liquidi e cibo a poche ore dalla conclusione dell’intervento, conservando, inalterata, la canalizzazione intestinale). Questo approccio risulta particolarmente vantaggioso nei feocromocitoma bilaterale, in quanto il malato, preparato sul lettino operatorio in posizione prona, si trova già posto in modo corretto per entrambe le surrenectomie. In questi casi l’intervento può essere eseguito contemporaneamente da due équipe distinte, ciascuna impegnata nell’exeresi della ghiandola da un lato (surrenectomia bilaterale simultanea), con netta riduzione dei tempi operatori e della durata dell’anestesia.
Surrenectomia “single–site”. Sempre nell’ottica di una riduzione del trauma operatorio, è stata recentemente introdotta una variante tecnica, che prevede l’esecuzione dell’intervento con un’unica incisione cutanea della lunghezza di 2 cm, o a livello addominale o in sede lombare, attraverso la quale vengono introdotti gli strumenti chirurgici (la tecnica viene definita “single port adrenalectomy” se per via addominale o “SARA, single access retroperitoneoscopic adrenalectomy” per la via posteriore). Le indicazioni sono limitate ai pazienti magri (BMI 20–24), con minima quantità di grasso peri-renale e con feocromocitomi di piccole dimensioni.
Chirurgia robotica. La chirurgia robotica ha trovato un interessante campo di applicazione anche nella surrenectomia mini-invasiva. La procedura chirurgica è sostanzialmente la stessa, ma a differenza delle tecniche precedentemente descritte il chirurgo è seduto alla console, lontana dal tavolo operatorio, dalla quale controlla il movimento delle braccia meccaniche del robot alle quali sono collegati gli strumenti chirurgici. I vantaggi di questa tecnologia sono soprattutto ad appannaggio del chirurgo, che ha una visione tridimensionale del campo operatorio, utilizza strumenti dotati di grande ampiezza di movimenti e di stabilità (viene eliminato il tremore umano), che consentono di compiere gesti chirurgici molto accurati, estremamente precisi, particolarmente utili nella fase più delicata dell’intervento, l’isolamento della vena media. I risultati della surrenectomia robotica sono sovrapponibili a quelli degli altri interventi mini-invasivi; al contrario, i costi del robot e della manutenzione degli strumenti sono molto elevati, tanto da limitare il suo impiego a casi selezionati.
Risultati, decorso e complicanze
Nonostante i feocromocitomi si presentino con maggior sviluppo della rete vascolare rispetto ad altre affezioni benigne del surrene, le perdite ematiche intra-operatorie sono in genere modeste e tali da non rendere necessario l’impiego di emotrasfusioni. Nel decorso post-operatorio degli interventi mini-invasivi, l’alimentazione per os viene ripresa in prima giornata post-operatoria (con la tecnica retro-peritoneale anche dopo poche ore dalla conclusione dell’intervento). La dimissione avviene in seconda-terza giornata.
Il trattamento chirurgico presenta alte percentuali di successo, con mortalità pressoché nulla e con incidenza di complicanze estremamente contenuta. Le più frequenti sono l’emorragia (1.5–2%), che nella maggior parte dei casi non necessita di reintervento, e, nell’approccio mini-invasvo, lo sviluppo di laparocele in corrispondenza dell’accesso dei trocar nella parete addominale (0.5–1%) o la comparsa, con accesso retro-peritoneale, di una relaxatio della parete addominale in corrispondenza dei fianchi, per lesione traumatica, in genere reversibile, dell’XI nervo intercostale (2-3%).
SURRENECTOMIA PARZIALE “CORTICAL-SPARING”
Nei primi anni del Novecento studi sperimentali condotti sul modello animale (suini) hanno dimostrato la possibile sopravvivenza degli animali privati chirurgicamente dei 7/8 del tessuto ghiandolare surrenalico; nel 1936 e nel 1946 sono stati descritti in letteratura casi di pazienti sottoposti con successo a surrenectomia parziale bilaterale (5). Nonostante questo incoraggiante indirizzo di ricerca, fino agli anni Novanta la surrenectomia totale, mono o bilaterale, ha rappresentato il trattamento di scelta del feocromocitoma nell’ambito della MEN-2. Indubbiamente questo tipo di intervento offre garanzie assolute di cura, annullando o limitando a casi sporadici il rischio di recidiva, ma impone al paziente, nel caso di un’exeresi bilaterale, un trattamento steroideo sostitutivo definitivo, con il possibile sviluppo di osteoporosi e con il rischio di incorrere, nel 20-30% dei casi, in una crisi addisoniana, talvolta con esito infausto (6, 7, 8).
Alla luce di queste problematiche, la surrenectomia parziale “cortical-sparing” rappresenta l’intervento ideale per la cura del feocromocitoma nell’ambito di una MEN-2, in quanto prevede l’asportazione completa della lesione della midollare, mantenendo in sede una porzione di corticale sana, ben vascolarizzata, in grado di garantire una sintesi adeguata di ormoni steroidei. L’exeresi del feocromocitoma deve essere completa, senza interruzioni della capsula, per evitare il rischio di recidiva locale. Sotto il profilo oncologico, l’enucleazione del feocromocitoma è inoltre giustificata dalla natura benigna della malattia, che solo eccezionalmente manifesta nel follow-up il carattere di malignità.
Con l’avvento della laparoscopia la surrenectomia “cortical-sparing” è stata riproposta con successo e si sta progressivamente diffondendo nel trattamento chirurgico non solo della MEN-2, ma anche di altre malattie ereditarie del surrene (sindrome di Von Hippel-Lindau, neurofibromatosi). La tecnologia abbinata alla chirurgia endoscopica ha infatti favorito lo sviluppo di questa procedura conservativa: l’ingrandimento dell’immagine ottenuto con la videocamera consente di evidenziare con maggior precisione il “piano di clivaggio” tra il feocromocitoma e la porzione di corticale da preservare; inoltre, gli strumenti di coagulazione e taglio a ultrasuoni o a radiofrequenza utilizzati in laparoscopia consentono di sezionare la ghiandola, peraltro riccamente vascolarizzata, ottenendo un’emostasi accurata e stabile (9, 10).
La pianificazione dell’intervento viene stabilita sulla base delle informazioni iconografiche fornite da TC ed RM. Questi stessi strumenti di indagine, utilizzati anche per la valutazione del volume di ghiandola residuo ad un intervento di “sparing”, hanno permesso di stabilire che la conservazione del 15% del parenchima ghiandolare (pari ad 1/3 del volume di uno dei due surreni) è sufficiente per assicurare al paziente un’autonomia funzionale (5, 7).
I risultati della tecnica “cortical-sparing” riportati in letteratura sembrano molto favorevoli: più del 90% dei pazienti operati per forme bilaterali presenta piena autonomia dalla terapia sostitutiva steroidea, con una percentuale di recidiva < 10% nel surrene trattato conservativamente (5). Nelle recidive è possibile reintervenire chirurgicamente, spesso con tecnica endoscopica, per “totalizzare” la surrenectomia, nel lato in cui si è riproposta la malattia.
CONCLUSIONI
La terapia chirurgica costituisce l’unica possibilità di cura definitiva del feocromocitoma insorto nell’ambito di una MEN-2. L’intervento chirurgico viene oggi condotto nella maggioranza dei casi con tecnica endoscopica mini-invasiva, trans- o retro-peritoneale, con alte percentuali di successo e con incidenza trascurabile di complicanze. La surrenectomia “cortical-sparing” si sta imponendo come l’intervento di scelta, in quanto garantisce al paziente un’autonomia funzionale surrenalica nel 90% dei casi, con percentuali di recidiva ipsilaterale al surrene trattato < 10%. La procedura conservativa deve essere pianificata in tutti i casi di feocromocitoma nell’ambito di una MEN-2, anche nelle forme ad esordio monolaterale, per l’alta probabilità che la malattia si riproponga, anche a distanza di molti anni, nel surrene controlaterale.
BIBLIOGRAFIA
- Rodriguez JM, Balsalobre M, et al. Pheochromocytoma in MEN 2A syndrome: study of 54 patients. World J Surg 2008, 32: 2520-6.
- Zuluaga-Gomez A, Arrabal-Polo MA, Arrabal-Martin M, Lahoz Garcia C. Management of pheochromocytoma during pregnancy: laparoscopic adrenalectomy. Am Surg 2012, 78: 156-8.
- Iusco D, Sarli L, Di Mauro D, et al. Endoscopic treatment of bilateral pheochromocytoma in MEN 2A syndrome: case report and review of the literature. G Chir 2007, 28: 363-6.
- Boniardi M. Chirurgia mini-invasiva del surrene: quali novità? AME flash n. 11 – maggio 2013.
- Alesina PF, Hinrichs J, Maier B, et al. Minimally invasive cortical-sparing surgery for bilateral pheochromocytoma. Langenbeck’s Arch Surg 2012, 397: 233-8.
- Scholten A, Valk GD, Ulfman D, et al. Unilateral subtotal adrenalectomy for pheochromocytoma in MEN type 2 patients. Ann Surg 2011, 254: 1022-7.
- Grubbs EG, Rich TA, Ng C, et al. Long-term outcomes of surgical treatment for hereditary pheochromocytoma. Am Coll Surg 2013, 216: 280-9.
- Azari R, Scheuba C, Kaczirck K, Niederle B. Estimated risk of pheochromocytoma recurrence after adrenal-sparing surgery in patients with Multiple Endocrine Neoplasia type 2A. Arch Surg 2006, 141: 1199-205.
- Cavallaro G, Letizia C, Polistena A, De Toma G. Laparoscopic adrenal-sparing surgery: personal experience, review on technical aspects. Updates Surg 2011, 63: 35-8.
- Neumann HPH, Bender BU, Reincke M, et al. Adrenal-sparing surgery for pheochromocytoma. Br J Surg 1999, 86: 94-7.
Terapia dell'iperparatiroidismo nell'ambito della MEN-2
Farmacologica: non differisce da quella dell'iperparatiroidismo primario
Terapia chirurgica per l'iperparatiroidismo nell'ambito della MEN-2: quando e quale?
Giancarlo Pansini & Simona Bonazza
Sezione di Clinica Chirurgica, Dipartimento di Morfologia, Chirurgia e Medicina Sperimentale, Università degli Studi di Ferrara
Introduzione e premesse
Nella MEN-2A, i pazienti hanno una predisposizione genetica a una patologia multi-ghiandolare, una certa frequenza di ghiandole paratiroidi sovrannumerarie o ectopiche, nonché un’incidenza non trascurabile di iperparatiroidismo (IPT) persistente o recidivo dopo ogni trattamento chirurgico (1,2). Nell’era della diagnosi genetica della MEN-2A molti bambini e adolescenti vengono sottoposti precocemente a tiroidectomia per il CMT, nella prima o seconda decade di vita, quando invece l’alterazione paratiroidea, intesa come ingrandimento ghiandolare, è raramente presente.
Alla luce di tali dati, il trattamento delle paratiroidi in questa sindrome rimane tuttora controverso (3,4).
Quando operare?
La decisione di sottoporre a intervento un paziente con MEN-2A è necessariamente ponderata sulla scorta delle conoscenze acquisite sulla storia naturale dell’IPT nella MEN-2A, nonché sul rischio di IPT persistente o recidivo dopo chirurgia, di ipocalcemia permanente post-operatoria e sui rischi di complicazioni chirurgiche di fronte alla necessità di interventi reiterati in caso di persistenza o recidiva dell’IPT. In linea di massima, le indicazioni alla paratiroidectomia e i criteri diagnostici pre-operatori sono sovrapponibili a quelli riconosciuti nella MEN-1 e nell’IPT sporadico; la maggior parte dei pazienti operati ha sintomi evidenti come dolore osseo o derivati dalla presenza dei calcoli renali o sintomi più aspecifici, dalla facile affaticabilità fino alla letargia, legati all’ipercalcemia (4-6).
Quale tipo di intervento? Paratiroidectomia parziale, subtotale o totale?
La modalità ideale del trattamento chirurgico delle paratiroidi nei pazienti con IPT e MEN-2A deve tenere conto di alcuni aspetti della malattia nell’ambito della sindrome:
- le modalità di comparsa della malattia e le trasformazioni patologiche delle paratiroidi sono variabili e non esattamente sovrapponibili a quelle conosciute nella MEN-1 (nella maggior parte dei pazienti con MEN-2A le paratiroidi ingrandite sono meno di quattro);
- i risultati dei diversi trattamenti sono spesso contraddittori (5).
Nonostante che nella MEN-2A non tutte le ghiandole siano sempre ingrandite, la procedura iniziale prevede innanzitutto l’esplorazione cervicale bilaterale, possibilmente con identificazione di tutte le quattro paratiroidi. In questa prospettiva, la paratiroidectomia cosiddetta mini-invasiva, non viene raccomandata perché non consente l’identificazione routinaria efficace e sicura di tutte le paratiroidi. In seguito, in relazione (o indipendentemente) alla conformazione patologica delle paratiroidi identificate di ogni singolo paziente, si stabilisce quale estensione riservare all’asportazione delle paratiroidi. Alcuni gruppi sostengono che la paratiroidectomia parziale o selettiva, quindi comprensiva delle sole paratiroidi ingrandite (qualora non lo fossero tutte e quattro) sembrerebbe dimostrarsi adeguata nei pazienti con malattia non palesemente multi-ghiandolare. Altri esperti raccomandano invece la paratiroidectomia totale con auto-trapianto eterotopico (nell’avambraccio) di frammenti ghiandolari opportunamente preparati, particolarmente in quei pazienti che precocemente, sulla scorta della diagnosi genetica, vanno incontro alla tiroidectomia totale per il CMT. Anche in questa procedura, l’impiego dell’auto-trapianto paratiroideo nei muscoli dell’avambraccio è destinato a semplificare il controllo ed il trattamento dell’IPT persistente o recidivo, in caso di iperfunzione del residuo paratiroideo, poiché anche in questo caso, l’accesso all’avambraccio è enormemente facilitato rispetto al collo, consentendo la rimozione parziale o totale del tessuto trapiantato, se necessario.
Le ragioni a favore della procedura radicale sono molteplici:
- in presenza di un difetto genetico, l’alterazione patologica è e sarà potenzialmente multi-ghiandolare;
- l’IPT è frequentemente persistente o recidivo dopo procedure conservative;
- la procedura totale con auto-trapianto minimizza le conseguenze dell’ipoparatiroidismo post-operatorio potenzialmente causato dalle manovre durante la tiroidectomia per il CMT;
- previene i rischi delle complicazioni che potrebbe comportare un nuovo accesso cervicale per la paratiroidectomia in quei pazienti precedentemente già sottoposti alla tiroidectomia per il CMT, che fossero andati incontro, nel corso della loro vita, alla comparsa tardiva dell’IPT.
Le principali critiche alla paratiroidectomia totale sono:
- una procedura così estesa appare eccessivamente radicale per un’alterazione paratiroidea che è solo relativamente frequente, che non sempre è estesa macroscopicamente a tutte le quattro paratiroidi;
- che i risultati a distanza della paratiroidectomia parziale o selettiva rappresentano un’eccellente cura per i pazienti con ingrossamenti limitati a meno di quattro ghiandole;
- l’incidenza dell’ipoparatiroidismo anche dopo auto-trapianto è comunque inaccettabile nella maggioranza dei pazienti, nei quali la complicazione della terapia rappresenta addirittura un risultato peggiore della malattia iniziale (7-11).
Il principale studio retrospettivo fra pazienti con MEN-2A sottoposti a paratiroidectomia confronta i risultati fra la procedura selettiva o subtotale e quella totale con auto-trapianto, dichiarando una percentuale di successo nei due gruppi pari all’88% e 82%, rispettivamente, nel corso di un follow-up di 8 anni. Nel gruppo trattato con paratiroidectomia parziale, i casi di IPT recidivante o persistente sembrerebbero causati dalla presenza di ghiandole ectopiche o sovrannumerarie, nonché dallo sviluppo di neoplasia nel tessuto residuo. Tuttavia, in entrambi i gruppi la percentuale di ipoparatiroidismo permanente rimane significativa, con un aumento importante nel caso di pazienti sottoposti a re-intervento per IPT recidivante o persistente (12).
In sintesi, l’approccio ideale al trattamento chirurgico dei pazienti con MEN-2A e IPT rimane controverso. La maggior parte delle argomentazioni deporrebbe più a favore di una paratiroidectomia parziale, selettiva oppure subtotale (3 ghiandole + ½ della quarta paratiroide), con sorveglianza clinica del possibile IPT recidivo e trattamento cronico dell’eventuale ipoparatiroidismo permanente. A questa linea si attiene l’esperienza dell’autore.
Bibliografia
- Carney JA. Familial multiple endocrine neoplasia syndromes: components, classification, and nomenclature. J Int Med 1998, 243: 425–32.
- Wohllk N, Schweizer H, Erlic Z, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab 2010, 24: 371–87.
- Carney JA. Familial multiple endocrine neoplasia: the first 100 years. Am J Surg Pathol 2005, 29: 254–74.
- Romei C, Pardi E, Cetani F, et al. Genetic and clinical features of multiple endocrine neoplasia type 1 and 2. J Oncol 2012, 2012: 705036.
- Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN Type 1 and Type 2. J Clin Endocrinol Metab 2001, 86: 5658-71.
- Callander GG, Rich TA, Terrier ND. Multiple endocrine neoplasia syndromes. Surg Clin N Am 2008, 88: 863-95.
- Dalbridge LW, Younes NA, Guinea AL, et al. Surgery for primary hyperparathyroidism 1962-1996; indications and outcomes. Med J Aust 1998, 168: 153-6.
- Freier DT, Thompson NW, Sisson JC, et al. Dilemmas in the early diagnosis and treatment of multiple endocrine adenomatosis, type II. Surgery 1977, 82: 407-13.
- O’Riordain DS, O’Brien T, Grant CS, et al. Surgical management of primary hyperparathyroidism in multiple endocrine neoplasia type 1 and 2. Surgery 1993, 114: 1031-9.
- Wells SA, Farndon JR, Dale JK, et al. Long-term evaluation of patients with primary parathyroid hyperplasia managed by parathyroidectomy and heterotopic autotransplantation. Ann Surg 1980, 192: 451-8.
- Olson JA, De Benedetti MK, Baumann DS, et al. Parathyroid autotransplantation during thyroidectomy: results of long-term follow-up. Ann Surg 1996, 223: 472-80.
- Raue F, Krainps JL, Dralle H, et al. Primary hyperparathyroidism in multiple endocrine neoplasia type 2A. J Intern Med 1995, 238: 369-73.
Follow-up della MEN-2
Alberto Falchetti1 & Nadia Cremonini2
1Centro Hercolani, Bologna; 2UOSD di Endocrinologia – Azienda USL di Bologna
I pazienti MEN-2 devono essere seguiti con attento follow-up delle patologie per le quali sono già stati trattati (carcinoma midollare, MTC, feocromocitoma, FCC, e/o iperparatiroidismo primitivo, IPTH), ma poiché tali patologie possono manifestarsi in momenti diversi, il follow-up si embrica di necessità con lo screening.
Il follow-up non si differenzia da quello effettuato per le forme sporadiche.
Carcinoma midollare tiroideo (MTC)
In circa il 50% dei soggetti con diagnosi di MTC, sottoposti a tiroidectomia totale (non preventiva come nei portatori di mutazione germinale di RET) e dissezione dei linfonodi del collo, si verifica recidiva di malattia (1); inoltre, negli individui con mutazione germinale RET e normali livelli plasmatici basali di calcitonina (Ct) è frequente il riscontro di microfocolai di MTC (2,3). Pertanto, si raccomanda il monitoraggio continuo dei pazienti o per confermare la guarigione o per seguire i pazienti con malattia residua o recidiva.
Il follow-up si fonda sulla determinazione di Ct e di CEA, da effettuarsi dopo due-tre mesi dall’intervento, in associazione a controllo ecografico del collo; la maggioranza dei componenti della Task Force delle Linee Guida ATA non ritiene indicato ricorrere a test di stimolo in presenza di Ct indosabile (< 2 pg/mL), in quanto se la Ct è elevata solo dopo stimolo, la quota di tessuto neoplastico è talmente esigua da non essere rilevata con le tecniche di immagine (4); nella pratica clinica questo è stato confermato ampiamente. Di avviso diverso sono, invece, alcuni esperti europei, che ritengono indicato effettuare test di stimolo per la condizione sopra descritta: in presenza di Ct indosabile anche dopo stimolo (remissione biochimica completa), il rischio di recidiva biochimica è comunque del 3% circa (5).
La tempistica delle determinazioni successive di Ct e CEA e della diagnostica per immagini è dettata dai livelli post-operatori dei marcatori: se indosabili, prevede una misura annuale di Ct nel siero (4). In presenza di malattia residua, la tempistica dei controlli biochimici, clinici e di imaging dipende dai livelli di Ct, che correlano con l’estensione della malattia stessa (follow-up di MTC sporadico).
Necessario anche il monitoraggio dell’adeguatezza della terapia sostitutivacon levo-tiroxina, da effettuarsi con determinazione di TSH Reflex ogni 3-6 mesi inizialmente, indi, raggiunto il compenso ottimale, con controlli annuali.
Feocromocitoma
Il follow-up di FCC nelle MEN-2A e MEN-2B non ha come scopo principale l’individuazione di secondarismi (le forme maligne sono rarissime), bensì quello di porre:
- diagnosi precoce di malattia nel surrene controlaterale nei pazienti con malattia monolaterale all’esordio e trattati con surrenectomia monolaterale: particolare attenzione va prestata ai pazienti MEN-2B e ai pazienti MEN-2A con mutazione RET c634 che presentano elevata percentuale di FCC bilaterale;
- diagnosi di persistenza di malattia o recidiva nei pazienti con FCC bilaterale all’esordio e trattati con tecnica “cortical-sparing”, anche se le percentuali riportate in letteratura sono veramente molto basse (6).
Il monitoraggio va effettuato con determinazione di metanefrine plasmatiche o urinarie, da eseguirsi dopo 3 mesi dall’intervento e, se nella norma e il paziente è asintomatico, ogni sei mesi per i tre anni successivi e a seguire annualmente (7).
Nei pazienti sottoposti a surrenectomia bilaterale tradizionale va inoltre controllata l’adeguatezza della terapia sostitutiva per l’insufficienza surrenalica; nei pazienti trattati con tecnica “cortical-sparing” che non necessitano di terapia sostitutiva va monitorata la funzione cortico-surrenalica.
I pazienti devono essere sottoposti a periodici controlli clinici, con tempistica diversa, in base alla sintomatologia e al quadro biochimico rilevato.
Iperparatiroidismo primitivo
Nella MEN-2A il trattamento chirurgico per IPTH prevede l’exeresi della/e paratiroide/i aumentata/e di dimensioni (8). Il follow-up è volto alla verifica della guarigione dell’IPTH, e va effettuato mediante determinazione della calcemia totale, fosforemia e PTH; se a distanza di sei mesi dall’intervento il mantenimento di una normocalcemia ci permette di stabilire la guarigione, successivamente i controlli biochimici vanno effettuati annualmente, per lo screening della malattia nelle paratiroidi residue.
Nei rari casi che richiedono paratiroidectomia totale e autotrapianto di frammenti paratiroidei, si rende necessario il monitoraggio annuale di calcemia e PTH per un possibile ipoparatiroidismo. È consigliabile anche determinare i livelli di vitamina D.
Bibliografia
- Cohen MS, Moley JF. Surgical treatment of medullary thyroid carcinoma. J Intern Med 2003, 253: 616-26.
- Skinner MA, DeBenedetti MK, Moley JF, et al. Medullary thyroid carcinoma in children with multiple endocrine neoplasia types 2A and 2B. J Pediatr Surg 1996, 31: 177-81.
- Elisei R, Romei C, Renzini G, et al. The timing of total thyroidectomy in RET gene mutation carriers could be personalized and safely planned on the basis of serum calcitonin: 18 years experience at one single center. J Clin Endocrinol Metab 2012, 97: 426-35.
- Kloos RT, Eng C, Evans DB, et al. American Thyroid Association Guidelines Task Force. Medullary thyroid cancer: management guidelines of the American Thyroid Association. Thyroid 2009, 19: 565-612.
- Machens A, Schneyer U, Holzhausen HJ, et al. Prospect of remission in medullary thyroid carcinoma according to basal calcitonin level. J Clin Endocrinol Metab 2005, 90: 2029-34.
- Alesina PF, Hinrichs J, Achmid KW, et al. Minimally invasive cortical-sparing surgery for bilateral pheochromocytomas. Langenbecks Arch Surg 2012, 397: 233-8.
- Kulke MH, Benson AB, Bergsland E, et al. NCCN Clinical Practice Guidelines in Oncology. Neuroendocrine tumors. Version 1.2013 – NCCN.org
- Zini M, Attanasio R, Cesareo R, et al. AME position statement: primary hyperparathyroidism in clinical practice. J Endocrinol Invest 2012, 35 (Suppl 7): 1–21.
MEN-4
Irene Gagliardi e Anna Crociara
Sezione di Endocrinologia, Azienda Ospedaliera-Universitaria S. Anna, Ferrara
(aggiornato al 3 marzo 2020)
Clinica
La sindrome MEN-4 è caratterizzata clinicamente dalla presenza di neoplasie simili al quadro sindromico della MEN-1: tumori delle paratiroidi, tumori dell’ipofisi e neoplasie neuroendocrine pancreatiche, pur in assenza di mutazioni inattivanti a carico della porzione codificante del gene MEN-1 (1). Inoltre, sono state riportate associazioni con tumori renali, surrenalici, intestinali, degli organi riproduttivi, lipomi e meningiomi, carcinoidi bronchiali, carcinoma papillare tiroideo (2-6).
La manifestazione clinica più frequente (80% dei pazienti) è la neoplasia delle paratiroidi associata a iperparatiroidismo primario (7). Generalmente è diagnosticata in individui con almeno 40 anni di età (3); la diagnosi più precoce è avvenuta in un soggetto di 15 anni (8).
La seconda manifestazione più comune sono gli adenomi ipofisari (37% dei pazienti), che generalmente sono più piccoli e meno aggressivi rispetto agli adenomi ipofisari MEN-1 associati (7): sono stati registrati casi di GHoma, ACTHoma, adenoma non funzionante, PRLoma, con un’età di insorgenza compresa tra 30 e 79 anni (7).
I NET pancreatici, incluso il gastrinoma, sono stati osservati nelle MEN-4 con una prevalenza di circa il 25% e una penetranza ridotta rispetto alla MEN-1 (2,3,7).
Genetica ed epidemiologia
La sindrome MEN-4 trova la sua origine da una variante delle sindromi MEN, che spontaneamente si sviluppava nel ratto, denominata MENX (9). Gli animali colpiti sviluppavano molteplici tumori endocrini, con uno spettro di caratteristiche cliniche comuni alle sindromi umane MEN-1 e MEN-2 (10). Studi genetici hanno identificato la causa della sindrome MENX nella mutazione germinale del gene CDKN1B.
Nell’uomo la sindrome è causata da mutazioni germinali in eterozigosi di CDKN1B (cyclin-dependent kinase inhibitor 1B), gene localizzato sul cromosoma 12p13, che codifica per p27, una proteina coinvolta nella regolazione del ciclo cellulare. Tali mutazioni influenzano la localizzazione cellulare e la stabilità di p27, ma anche il legame con partner funzionali come Cdk2 (chinasi-ciclina dipendente di tipo 2) o Grb2 (proteina legante il recettore dei fattori di crescita tipo 2) (1). Ad oggi, ci sono 17 mutazioni germinali di CDKN1B identificate e riportate in letteratura, che spiegano l’1.5-3.7% dei pazienti con MEN negativi per mutazioni di menin (7,11-14), senza una chiara correlazione genotipo-fenotipo.
Per quanto riguarda le modalità di segregazione della mutazione, i dati sono ancora scarsi. Pellegata et al (9) suggerivano una modalità di trasmissione autosomica dominante, descrivendo il caso di una variante patogenetica di CDKN1B riscontrata in due sorelle con fenotipo clinico MEN-1 simile. L’analisi dell’aplotipo mostrava che l’allele mutato era stato ereditato dal padre, il quale a sua volta aveva ricevuto diagnosi di acromegalia. La trasmissione autosomica dominante della mutazione è stata recentemente supportata anche dalla scoperta in 13 membri di una numerosa famiglia di una nuova mutazione di CDKN1B, segregata con le manifestazioni cliniche in due generazioni successive (3).
Tutti i casi riportati in letteratura di adulti portatori di una mutazione di CDKN1B avevano una patologia MEN-4 relata, suggerendo una penetranza completa della patologia (3).
Nel gene umano CDKN1B sono stati identificati diversi polimorfismi a singolo nucleotide (SNPs), tre dei quali sembrano essere potenzialmente funzionali:
- -838C>A (rs36228499);
- -79C>T (rs34330);
- -326T>G (V109G, rs2066827).
Studi epidemiologici hanno identificato un’associazione significativa tra lo SNP codificante rs2066827 (V109G) e il cancro della prostata, della mammella e il carcinoma squamoso del cavo orale (15). Il ruolo di questo polimorfismo nell’aumentare il rischio neoplastico rimane ancora controverso, dal momento che i risultati di diversi studi non sono sempre coerenti. Recentemente lo SNP -79C>T (rs34330) è stato associato al rischio di sviluppare il carcinoma papillare tiroideo variante follicolare (16). Uno studio ha riscontrato che il polimorfismo CDKN1B rs2066827 potrebbe essere associato ad una ridotta suscettibilità al carcinoma ovarico (17).
Diagnostica e gestione
Considerata la scarsità di casi descritti in letteratura, non esistono ad oggi linee guida e/o raccomandazioni per la gestione della sindrome MEN-4. Nella pratica clinica si suggerisce di ricercare le mutazioni di CDKN1B in individui con un fenotipo MEN-1 simile ma negativi per mutazioni del gene menin. In caso di positività di mutazione di CDKN1B, lo screening dovrebbe essere esteso successivamente anche ai familiari di primo grado, con o senza manifestazioni cliniche (7).
È consigliato sottoporre a indagine genetica per la ricerca di mutazioni del gene CDKN1B non solo pazienti con fenotipo MEN-1-simile, ma anche pazienti in cui è presente una predisposizione allo sviluppo di adenomi ipofisari e di adenomi delle paratiroidi sporadici.
La diagnosi molecolare della MEN-4, eseguita attraverso la ricerca di mutazioni nel DNA genomico estratto da sangue periferico, prevede amplificazione mediante PCR (reazione a catena della polimerasi) della regione 5’UTR (untraslated region), degli esoni 1, 2 e 3 e della regione 3’UTR del gene CDKN1B. I prodotti PCR così ottenuti vengono successivamente sottoposti a sequenziamento automatico (metodo Sanger). Le sequenze di DNA, rappresentate dagli elettroferogrammi prodotti, vengono allineate con la sequenza normale del gene alla ricerca di mutazioni.
Bibliografia
- Ozawa A, Agarwal SK, Mateo CM, et al. The parathyroid/pituitary variant of multiple endocrine neoplasia type 1 usually has causes other than p27 Kip1 J Clin Endocrinol Metab 2007, 92: 1948–51.
- McDonnell JE, Gild ML, Clifton‐Bligh RJ, Robinson BG. Multiple endocrine neoplasia: an update. Intern Med J 2019, 49: 954–61.
- Frederiksen A, Rossing M, Hermann P, et al. Clinical features of multiple endocrine neoplasia type 4: novel pathogenic variant and review of published cases. J Clin Endocrinol Metab 2019, 104: 3637–46.
- Malanga D, De Gisi S, Riccardi M, et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol 2012, 166: 551–60.
- Occhi G, Regazzo D, Trivellin G, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS Genet 2013, 9: e1003350.
- Tonelli F, Giudici F, Giusti F, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol 2014, 171: K7–17.
- Alrezk R, Hannah-Shmouni F, Stratakis CA. MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer 2017, 24: T195–208.
- Elston MS, Meyer-Rochow GY, Dray M, et al. Early onset primary hyperparathyroidism associated with a novel germline mutation in CDKN1B. Case Rep Endocrinol 2015, 2015: 510985.
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA 2006, 103: 15558–63.
- Fritz A, Walch A, Piotrowska K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res 2002, 62: 3048–51.
- Vandeva S, Daly AF, Petrossians P, et al. Somatic and germline mutations in the pathogenesis of pituitary adenomas. Eur J Endocrinol 2019, 181: R235–
- Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 2009, 94: 1826–34.
- Georgitsi M, Raitila A, Karhu A, et al. Germline CDKN1B /p27 Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab 2007, 92: 3321–5.
- Molatore S, Marinoni I, Lee M, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: clinical, genetic and functional characterization. Hum Mutat. 2010, 31: E1825–35.
- Wei F, Xu J, Tang L, et al. p27 Kip1 V109G polymorphism and cancer risk: a systematic review and meta-analysis. Cancer Biother Radiopharm 2012, 27: 665–71.
- Landa I, Montero-Conde C, Malanga D, et al. Allelic variant at −79 (C>T) in CDKN1B (p27Kip1) confers an increased risk of thyroid cancer and alters mRNA levels. Endocr Relat Cancer 2010, 17: 317–28.
- Lu Y, Gao K, Zhang M, et al. Genetic association between CDKN1B rs2066827 polymorphism and susceptibility to cancer. Medicine (Baltimore) 2015, 94: e1217.
Sindrome di Von Hippel-Lindau
Marta Bondanelli
Sezione di Endocrinologia, Dipartimento di Scienze Mediche, Università degli Studi di Ferrara
DEFINIZIONE ED EPIDEMIOLOGIA
La sindrome di Von Hippel-Lindau (VHL) è una malattia a carattere ereditario, a trasmissione autosomica dominante, con elevata penetranza (80-90%), caratterizzata dall’associazione di varie neoplasie (tab 1):
- emangiobastomi del sistema nervoso centrale (SNC) (cerebellari e spinali);
- emangiomi retinici;
- carcinoma renale a cellule chiare (RCC) e cisti renali;
- feocromocitoma;
- tumori del sacco endo-linfatico dell’orecchio medio;
- tumori del pancreas;
- cistoadenoma papillare dell’epididimo o del ligamento largo.
| Tabella 1 | ||
| Manifestazioni | Età media (anni, range) | Frequenza |
| Sistema nervoso centrale | ||
| Emagioblastomi retinici | 25 (1-68) | 25-60% |
Emagioblastomi del SNC
|
30 (9-78) 33 (9-78) 32 (12-46) 33 (12-66) |
60-80% 44-72% 10-25% 13-50% < 1% < 1% |
| Tumori del sacco endo-linfatico | 22 (12-50) | 10-16% |
| Visceri | ||
| Carcinoma e cisti renali | 40 (13-70) | 25-70% |
| Feocromocitoma | 30 (5-58) | 10-20% |
| Neoplasie e cisti pancreatiche | 36 (5-70) | 35-77% |
| Cistoadenoma dell’epididimo | 25-60% | |
| Cistoadenoma del ligamento largo | (16-46) | raro |
L’età media di presentazione della sindrome è intorno ai 26 anni, con intervallo variabile dall’infanzia all’età adulta (1,2).
GENETICA E PATOGENESI
Il gene VHL è mappato sul cromosoma 3p25 e codifica per la proteina pVHL, responsabile della degradazione ubiquitina-mediata di numerose proteine cellulari. VHL si comporta come onco-soppressore, ossia un gene la cui normale funzione è quella di regolare la crescita cellulare (fig 1). Quando entrambe le copie di questo gene sono inattivate, la crescita cellulare è incontrollata e si sviluppano le neoplasie. In particolare, la carenza o inattività di pVHL causa stabilizzazione di HIF-1a (Hypoxia Induced Factor), con attivazione dei geni di risposta all’ipossia e conseguente aumento di processi metabolici, proliferazione cellulare, espressione di fattori angio-genetici e sintesi di eritropoietina. Una mutazione del gene VHL può inoltre causare alterazioni strutturali della matrice extra-cellulare e ridotta apoptosi cellulare (2,3,4).
Un’anomalia del gene VHL è presente in 1/36.000 nati vivi e può insorgere “de novo” nel 20% dei casi. Sono state descritte numerose mutazioni germinali del gene VHL, che causano delezioni (con rimozione di uno o più esoni), sostituzioni “missense” e mutazioni che conducono a proteine troncate (nonsense, indels, splice site), portando a fenotipi molto variabili (3,5).

Figura 1: Ruolo della proteina VHL sull’attivazione di HIF (modificato da 5)
La proteina pVHL (codificata dal gene VHL) forma un complesso stabile con altre proteine, che è responsabile della degradazione ubiquitina-mediata di numerose proteine cellulari. I principali bersagli del complesso pVHL, sono i fattori di trascrizione HIF 1 e 2.
La carenza o inattività di pVHL causa stabilizzazione di HIFa con attivazione dei geni di risposta all’ipossia e conseguente aumento di processi metabolici, sintesi di eritropoietina, espressione di fattori promuoventi angiogenesi e proliferazione cellulare.
HIF: Hypoxia Induced Factor; HPH: propyl idrossilasi; VEGF: Vascular endothelial growth factor; PDGF: Platelet-derived growth factor; TGFa: Transforming growth factor alpha; Glut1: Glucose transporter type1; EPO: eritropoietina
CLINICA, DIAGNOSI E TERAPIA
La malattia viene divisa in due fenotipi (tab. 2):
- tipo 1 con bassa probabilità di sviluppare feocromocitoma ed elevato rischio delle altre neoplasie;
- tipo 2 caratterizzata invece da feocromocitoma, che può manifestarsi da solo (2C), oppure associato a basso (2A) ed alto (2B) rischio di RCC.
Tali suddivisioni non sono tuttavia assolute, indicando la necessità di una continua sorveglianza per tutte le lesioni correlate alla sindrome anche nel fenotipo 2C (4,5).
Una mutazione germinale del gene VHL (R200 W) può anche causare una sindrome non tumorale, la policitemia di Chuvash, a trasmissione autosomica recessiva (5).
| Tabella 2 Fenotipi clinici e caratteristiche molecolari della sindrome di Von-Hippel Lindau |
|||
| Tipo | Manifestazioni cliniche | Tipo di mutazione | Attivazione HIF |
| 1 | Emangioblastomi della retina Emagioblastomi del SNC Carcinoma renale Neoplasie e cisti pancreatiche |
Delezione parziale o totale del gene o mutazione non senso | Importante |
| 2A | Feocromocitoma Emangioblastomi della retina Emagioblastomi del SNC |
Mutazione missenso | Importante |
| 2B | Feocromocitoma Emangioblastomi della retina Emagioblastomi del SNC Carcinoma renale Neoplasie e cisti pancreatiche |
Mutazione missenso | Importante |
| 2C | Feocromocitoma | Mutazione missenso | Meno importante o assente |
| I tumori del sacco endo-linfatico e i cistoadenomi sierosi dell’epididimo e del ligamento largo non sono stati assegnati a un fenotipo specifico | |||
In passato, la diagnosi di VHL veniva effettuata in presenza di almeno 2 manifestazioni tipiche; attualmente è indicata la conferma genetica mediante analisi su sangue periferico per la ricerca di una mutazione germinale VHL, specie nei pazienti con singola manifestazione (emangioblastomi retinici o del SNC). È possibile anche identificare soggetti con uno o più tumori tipici della sindrome, che non sono portatori di mutazione germinale VHL: in tal caso le manifestazioni possono essere legate a un mosaicismo somatico per mutazione intercorsa in più tessuti durante lo sviluppo embrionale. In caso di mutazione germinale nota, è possibile effettuare la diagnosi prenatale mediante amniocentesi o prelievo dei villi coriali, oppure una diagnosi genetica pre-impianto in caso di fertilizzazione in vitro (1)
Emangioblastomi del SNC
Rappresentano la più frequente (60-80% dei casi) ed emblematica manifestazione della sindrome, con età media di insorgenza intorno ai 30 anni (intervallo 9-70).
Si tratta di neoplasie benigne, frequentemente multiple, ben circoscritte, ricche di vasi con ampie cellule stromali, che non danno luogo né a invasione locale né a metastasi a distanza.
Si sviluppano più spesso nel cervelletto, midollo spinale, tronco cerebrale e radici nervose, e solo raramente al di sopra del tentorio.
La velocità di crescita è lenta, più spesso caratterizzata da periodi di quiescenza alternati a fasi di crescita; durante la gravidanza si può verificare una crescita rapida e la formazione di cisti con effetto massa.
Nelle localizzazioni sotto-tentoriali le lesioni possono presentarsi con sintomi di compressione delle strutture adiacenti, emorragia e/o ipertensione endocranica. La presenza di parestesie e/o dolore sono invece segni tipici iniziali di una lesione spinale.
Nel 20% dei casi l’emangiobastoma cerebellare si manifesta con policitemia, secondaria a produzione di eritropoietina.
La diagnosi si effettua mediante risonanza magnetica (RM), dove gli emangioblastomi si presentano con aspetto tipicamente macrocistico o talora come masse solide o miste.
Il trattamento è chirurgico. Una possibile alternativa, per le forme non cistiche di piccole dimensioni, è la radiochirurgia stereotassica, che causa riduzione della massa e blocco dalla crescita nel 90% dei casi, ma non previene la formazione di cisti, che possono richiedere un successivo trattamento chirurgico.
Gli emangioblastomi del SNC restano una delle principali cause di morbilità e mortalità di tali pazienti, data la loro molteplicità e la frequente insorgenza di nuove lesioni, per cui è indicato un screening con RM del capo (± colonna) ogni 12-36 mesi iniziando dall’adolescenza.Viene inoltre raccomandano di effettuare un’analisi del gene VHL in tutti i soggetti (o almeno in quelli di età < 50 anni) portatori di emangioblastomi retinici o del SNC, anche in presenza di lesione singola (1,2).
| Tabella 3 Screening raccomandato per i portatori della mutazione del gene VHL |
|||
| Tipo di tumore | Modalità di screening | Età di inizio | Frequenza |
| Angiomi retinici | Oftalmoscopia (diretta ed indiretta) | 2 anni | 12 mesi |
| Emangioblastoma del SNC | RM del capo (± colonna) | Adolescenza | 12-36 mesi |
| Carcinoma renale e tumori pancreatici | RM addome | 16 anni | 12 mesi |
| Feocromocitoma | Controllo pressione arteriosa Metanefrine urinarie e/o plasmatiche RM addome |
2 anni 8 anni (famiglie ad alto rischio) |
12 mesi 12 mesi |
| Tumori del sacco endo-linfatico | Valutazione audiologica | 5 anni | 2-3 anni (prima se clinicamente indicata) |
Emangiomi retinici
Spesso sono la prima manifestazione di VHL (età media 25 anni, intervallo 1-68), sono spesso multipli e bilaterali.
Il quadro istologico è quello di un emangioblastoma dei capillari retinici, con localizzazione periferica (85%) e/o nella regione iuxta-papillare (15%).
Tali lesioni possono dar luogo a complicanze emorragiche, glaucoma e distacco di retina, con rischio di cecità (25% dei casi).
Gli angiomi possono essere identificati con un esame indiretto del fondo oculare o tramite angioscopia con fluoresceina e possono essere trattati con successo mediante fotocoagulazione laser e/o crioterapia. In caso di localizzazione a livello del disco ottico, il trattamento può essere rischioso e viene riservato alle lesioni sintomatiche. Lesioni angiomatose severe possono inoltre prevedere trattamenti chirurgici vitreoretinici, al fine di impedire o ritardare la perdita di visus (1,2). In studi clinici, sono stati utilizzati farmaci anti-angiogenetici (ranibizumab) con risultati finora non soddisfacenti. Sono inoltre allo studio molecole target con attività inibente HIFa, il cui uso è prospettato non solo nel trattamento delle lesioni retiniche e del SNC, ma anche delle neoplasie renali (5).
Dato che l’identificazione di lesioni asintomatiche è solitamente in grado di prevenirne le complicanze, in tutti i portatori di anomalie del gene VHL è indicato lo screening oftalmologico annuale, iniziando dalla prima infanzia (1,2,4).
Carcinoma renale a cellule chiare (RCC)
È un’importante causa di morte nella VHL. Il rischio di sviluppare RCC è variabile nei diversi fenotipi, essendo del 70% nelle forme più comuni (tipo 1 e 2B). L’età media alla diagnosi è intorno ai 40 anni (intervallo 13-70).
RCC rimane spesso asintomatico per molti anni e diagnosticato incidentalmente in età avanzata; talora si manifesta con dolore o ematuria. È spesso bilaterale e multicentrico e preceduto da cisti renali multiple, alcune delle quali con significato pre-neoplastico.
La diagnosi è radiologica mediante TC o RM.
L’attuale approccio terapeutico prevede una regolare sorveglianza ai fini del risparmio della funzione renale, riservando la chirurgia (nefrectomia parziale) alle lesioni che raggiungono i 3 cm (RCC < 3 cm hanno basso potenziale metastatico e restano stabili a lungo). In caso di lesioni multiple o comparsa di nuove lesioni, possono essere utilizzate tecniche di ablazione percutanea con radiofrequenza e/o di crioablazione eco- o TC-guidata. La recente introduzione di terapie sistemiche con molecole target (sunitinib) può ridurre ulteriormente la necessità di nefrectomia bilaterale.
Lo screening per RCC va effettuato mediante RM dell’addome (o ecografia) ogni 12 mesi a partire dall’età di 16 anni (1,2,5).
Feocromocitoma
È presente nel 10-20% dei casi, con rischio nettamente maggiore nel fenotipo 2. L’età media di insorgenza è intorno ai 30 anni (intervallo 5-58 anni).
Può presentarsi in forma multipla e bilaterale e avere localizzazioni extra-surrenaliche (glomo carotideo, giugulare, tessuti peri-aortici). Il rischio di malignità è basso (< 5%). Si manifesta più spesso con sintomi tipici (ipertensione severa e/o intermittente, tachicardia, cefalea, sudorazioni, nausea, pallore, ecc), talora anche nell’infanzia. Può essere asintomatico nel 30% dei casi e può produrre solo norepinefrina.
La diagnosi si basa sul dato biochimico dell’eccesso di catecolamine e/o dei loro metaboliti misurati nel plasma o nelle urine delle 24 ore. La localizzazione si effettua con TC o RM, in associazione a tecniche di Medicina Nucleare (scintigrafia con MIBG, 18F-DOPA-PET, 18F-FDG-PET) per la diagnosi delle forme extra-surrenaliche e/o multiple.
Il trattamento è chirurgico, dopo adeguato alfa-blocco per evitare crisi ipertensive e seguendo opportuni protocolli di trattamento nel periodo peri-operatorio. Nelle forme bilaterali possono essere effettuate surrenectomie parziali con basso tasso di recidiva.
Lo screening deve essere effettuato annualmente a partire dalla prima infanzia (2 anni nelle famiglie ad alto rischio), mediante il dosaggio delle metanefrine urinarie o plasmatiche (queste ultime rappresentano il metodo più sensibile per la diagnosi di feocromocitoma in tale sindrome). Anche lo screening con metodiche di “imaging” va iniziato prima di quello previsto per RCC, specie nelle famiglie ad alto rischio (1,2,4).
Lesioni pancreatiche (tab 1)
Si osservano nel 35-70% dei portatori di VHL e comprendono cisti (70%), cistoadenomi sierosi (9-12%), emangioblastomi (>1%), adenocarcinomi (>1%) e tumori neuroendocrini (5-17%). Le cisti ed i cistoadenomi sono generalmente asintomatici e richiedono trattamento solo in caso di rare complicanze (pancreatiti, insufficienza pancreatica). Anche i tumori neuroendocrini sono spesso asintomatici, trattandosi di piccole lesioni non secernenti, ma possono anche dar luogo a sintomi di ipersecrezione ormonale (diarrea da VIP o crisi ipoglicemiche) e/o a metastasi a distanza. Il trattamento di tali lesioni è chirurgico. La chirurgia viene di solito riservata alle forme sintomatiche o, in assenza di sintomi, a quelle con diametro > 2 cm nella testa e >3 cm nel corpo o coda del pancreas (1,5,6).
Tumori del sacco endo-linfatico (tab 1)
Si riscontrano in corso di esami di “imaging” (CT o RM) nell'11-16% dei casi. Le forme bilaterali sono patognomoniche della malattia. Si tratta di lesioni benigne (cistoadenomi papillari) a crescita lenta, spesso asintomatiche. La manifestazione clinica più frequente è la perdita di udito, talora sono presenti tintinnii e vertigini, raramente paresi dei muscoli facciali. Il trattamento va considerato in relazione alla presenza e alla severità dei sintomi, date le possibili complicanze associate alla chirurgica. In tutti i portatori di VHL, è indicata una valutazione audiologica completa ogni 2-3 anni a partire dall’infanzia, riservando l’indagine RM in caso di sintomi o nel bambino con infezioni ripetute dell’orecchio medio (tab 3) (1,2,4).
Cisto-adenomi papillari dell'epididimo (tab 1)
sono presenti in oltre la metà dei maschi affetti da VHL e possono essere multipli e/o bilaterali. Si tratta di lesioni benigne, che originano dal dotto epididimale (derivante dal dotto mesonefritico embrionale), generalmente asintomatiche, rilevabili alla palpazione manuale e/o all’ecografia. La presenza di lesioni voluminose e bilaterali, oltre a creare discomfort, può compromettere la fertilità e richiedere un trattamento chirurgico (1,2).
Cistoadenomi papillari possono svilupparsi, seppur con minor frequenza, anche nelle giovani donne (16-46 anni) localizzandosi nel ligamento largo, ossia nei tessuti derivanti dal dotto mesonefritico embrionale (1,2).
BIBLIOGRAFIA
- Lonser RR, Glenn GM, Walther McC, et al. von Hippel-Lindau disease. Lancet 2003, 361: 2059–67.
- Maher ER, Neumann HPH, Richard S. von Hippel–Lindau disease: a clinical and scientific review. Eur J Hum Gen 2011, 19: 617–23.
- Richarda S, Gardiea B, Couvéa S, Gada S. Systemic VHL gene functions and the VHL disease. FEBS Lett 2012, 586: 1562–9.
- Barontini M, Dahia PLM. VHL Disease. Best Pract Res Clin Endocrinol Metab 2010, 24: 401–13.
- Richarda S, Gardiea B, Couvéa S, Gada S. Von Hippel–Lindau: How a rare disease illuminates cancer biology. Semin Cancer Biol 2013, 23: 26–37.
- Charlesworth M, Verbeke CS, Falk GA, et al. Pancreatic lesions in von Hippel–Lindau Disease? A systematic review and meta-synthesis of the literature. J Gastrointest Surg 2012, 16: 1422–8.
Neurofibromatosi tipo 1
Fabio Bondi, Maria Giulia Sama(*)
UOS Endocrinologia, (*)UOC Medicina Interna, Osp. Civile S. Maria delle Croci, Ravenna
Definizione, genetica e patogenesi
La neurofibromatosi tipo I (NF1) o malattia di Recklinghausen, è una sindrome genetica autosomico-dominante relativamente frequente, con prevalenza di un caso su 2500-3000 soggetti, indipendentemente da etnia e sesso, penetranza del 100% e grado variabile di espressione clinica (1).
La malattia è determinata da una mutazione germinale del gene NF1 (2), localizzato sul cromosoma 17q11.2, che codifica per la neurofibromina, proteina citoplasmatica lunga 2.818 aminoacidi, che agisce:
- inattivando p21Ras, proteina intra-cellulare importante per la regolazione della crescita e della sopravvivenza delle cellule, che a sua volta attiva molti importanti mediatori a valle, inclusi la proteina mTor;
- come modulatore positivo dei livelli di adenosin-monofosfato ciclico, effettore negativo della crescita cellulare.
La perdita della neurofibromina comporta quindi un aumento della proliferazione e della sopravvivenza cellulare. Le manifestazioni derivano da un difetto delle cellule originate dalla cresta neurale (cellule di Schwann, melanociti e forse fibroblasti endo-neurali), probabilmente come conseguenza dell'alterazione del fattore di crescita nervosa o del fattore di crescita gliale. L’elevata variabilità di espressione clinica non è presente solo nelle diverse famiglie, ma anche tra i diversi componenti affetti della medesima famiglia. Nei pazienti con mutazioni minori del gene non sono state dimostrate chiare correlazioni genotipo-fenotipo. Solo l’1-5% dei pazienti ha una delezione totale o subtotale del gene NF1: questi pazienti mostrano un fenotipo più grave, incluso l'inizio più precoce, un maggior numero di neurofibromi, aspetti dismorfici del volto, aumentato rischio di malignità e interessamenti del tessuto connettivo. È ancora controverso se questi pazienti abbiano un elevato rischio di sviluppo del tumore maligno della guaina dei nervi periferici (MPNST – malignant peripheral nerve sheath tumor), che può svilupparsi all’interno del neurofibroma plessiforme (3).
Clinica
Le caratteristiche cliniche della NF1 possono essere presenti alla nascita, ma spesso acquistano evidenza soltanto durante la tarda infanzia e l’adolescenza (1,2,4).
Nel 75% dei casi il disordine si caratterizza per la presenza di molteplici macchie caffè-latte e neurinomi cutanei; altri aspetti comuni sono i noduli di Lisch (amartomi melanocitici dell’iride, che non disturbano la visione) e le efelidi (ascellari o inguinali). In almeno il 50% delle persone con NF1 sono presenti difficoltà di apprendimento (1,4).
Altre manifestazioni meno comuni includono scoliosi, displasia tibiale, bassa statura, vasculopatie (coronaropatie, stenosi, aneurismi, malformazioni artero-venose), cardiopatie congenite (stenosi arteria polmonare nel 25%) e ipertensione. La stenosi dell'arteria renale è la causa più frequente di ipertensione, ma possono essere riscontrati coartazione aortica e feocromocitoma (2).
I pazienti con NF1 hanno un’aumentata suscettibilità ai tumori (rischio raddoppiato o quadruplicato rispetto alla popolazione generale), con un rischio di malignità stimata del 5-15%. Nel 25% dei pazienti si sviluppano neurofibromi plessiformi, gliomi del nervo ottico (i gliomi del nervo ottico a partenza chiasmatica possono determinare pubertà precoce) e altri gliomi del sistema nervoso centrale, tumori maligni della guaina dei nervi periferici (3,5), fibrosarcomi, tumori gastrointestinali stromali (5).
I tumori endocrini comprendono:
- feocromocitoma: i pazienti NF1 hanno una probabilità del 1-5% di sviluppare un feocromocitoma nell’arco della loro vita, che aumenta al 20-50% nei pazienti NF1 ipertesi; l’età media all’esordio è di 42 anni e per la maggior parte (90%) sono benigni e localizzati quasi esclusivamente a una o entrambe le ghiandole surrenali (2);
- NET duodeno-pancreatici: presenti nell’1% dei pazienti NF1; il somatostatinoma duodenale è il sottotipo più comune (2).
C’è un’associazione fra feocromocitoma e NET del duodeno, per cui la presenza di una patologia deve indurre a ricercare l'altra.
Le principali cause di decesso sono le malattie cardiovascolari e le neoplasie maligne che originano dalla guaina dei nervi.
Diagnosi
Usualmente la diagnosi si basa su almeno due dei seguenti sette aspetti clinici, stabiliti nel 1987 dal National Institutes of Health Consensus Development Conference (2):
- macchie caffe-latte (diametro maggiore > 5 mm nei prepuberi e > 15 mm dopo la pubertà);
- numerose aree iperpigmentate di 2-3 mm di diametro (efelidi) alle pieghe cutanee (ascella e inguine);
- noduli di Lisch (amartomi dell'iride);
- due o più neurofibromi o > 1 neurofibroma plessiforme;
- gliomi delle vie ottiche;
- lesioni ossee caratteristiche: displasia dello sfenoide, curvatura della tibia e della fibula, pseudo-artrosi degli arti inferiori, cifosi, scoliosi;
- familiarità di primo grado per NF1, diagnosticata secondo questi criteri.
La diagnosi differenziale si pone con le altre forme di neurofibromatosi (NF2 in primis) e con altre condizioni, soprattutto la sindrome Legius e la Constitutional Mismatch repair-deficiency syndrome.
Posta la diagnosi, i pazienti con NF1 debbono essere sottoposti a una serie di valutazioni, in modo che si possa stabilire la gravità della malattia e monitorarne la progressione:
- ai bambini devono essere controllati annualmente cute, rachide, vista e pressione arteriosa (2,4). Devono inoltre essere monitorati i loro progressi scolastici;
- in assenza di complicazioni, gli adulti affetti dalla NF1 richiedono un follow-up regolare da parte di un'equipe multidisciplinare.
Lo studio genetico nella NF1 è complesso, per il gran numero di possibili mutazioni in un gene di grandi dimensioni. Inoltre, nel 30-50% dei casi si tratta di nuove mutazioni.
Ove la familiarità ne ponga il sospetto, è possibile la diagnosi prenatale con villocentesi: il rischio per un paziente di avere un figlio gravemente affetto è di 1 su 12.
Terapie nei confronti delle cellule tumorali e della componente stromale
Il Tipifarnib è un inibitore dei processi di attivazione di RAS; studi di fase 1 hanno dimostrato buona tolleranza del farmaco in bambini con NF 1 con tumori solidi refrattari o con neurofibromi plessiformi.
La Rapamicina e i suoi analoghi, hanno azione inibitoria sulla via di segnalazione di mTor, e quindi vanno presi in considerazione nel trattamento dei tumori di questi soggetti (2).
AZD2171 e Talidomide sono farmaci che agiscono sulla vascolarizzazione del tumore (3), risultati efficaci nel trattamento dei tumori della guaina dei nervi periferici (5).
Il Pirfenidone è una sostanza anti-fibrotica che riduce le attività delle citochine, liberate dai fibroblasti in vicinanza dei neurofibromi, rendendo inefficace la rete cellulare peri-neoplastica di supporto (fibroblasti, mast-cellule e altri). È stato completato di recente uno studio di fase II in 24 pazienti con NF1 con neurinomi plessiformi: è stata dimostrata la stabilizzazione della malattia in 17 casi e la regressione in 3 di essi (3).
Bibliografia
- Riccardi VM. von Recklinghausen neurofibromatosis. N Eng J Med 1981, 305: 1617-27.
- Bartolozzi G. Neurofibromatosi tipo 1. Medico Bambino pagine elettroniche 2009, 12 (3).
- Widemann BC. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr Oncol Rep 2009, 11: 322-8.
- Williams VC, Lucas J, Babcock MA, et al. Neurofibromatosis type 1 revisited. Pediatrics 2009, 123: 124-33.
- Zehou O, Fabre E, Zelek L, et al. Chemotherapy for the treatment of malignant peripheral nerve sheath tumors in neurofibromatosis 1: a 10-year institutional review. Orphanet J Rare Dis 2013, 8: 127.