Il/la bambino/a normale

Asse GHRH-GH-IGF
Fabio Buzi
SC Pediatria, AO "C. Poma", Mantova
Il GH, il principale regolatore della crescita somatica, viene secreto in maniera pulsatile dalle cellule somatotrope dell’ipofisi anteriore. I regolatori della secrezione pulsatile del GH sono due fattori ipotalamici: il GHRH (GH-Releasing Hormone), stimolatorio, e la Somatostatina (GHRIF, GH-releasing – inhibiting hormone), inibitoria (1).
Il GHRH è un peptide di 44 aminoacidi che si trova in diverse forme circolanti, di cui le principali sono la 1-44 e la 1-40; per l’attività dell’ormone sono necessari almeno i primi 29 aminoacidi; tutte le forme derivano da modificazioni post-traduzionali di un più ampio peptide progenitore (2). Il GHRH agisce attraverso il legame a un recettore (GHRH-R) che fa parte della famiglia di recettori accoppiati a G-protein: il legame di GHRH al suo recettore incrementa i livelli di AMP ciclico intra-cellulare e attraverso una G-protein stimolatoria (Gs) attiva l’adenilato-ciclasi, incrementa il Ca libero intra-cellulare, rilascia GH preformato e stimola la trascrizione di GH mRNA e la sintesi di nuovo GH (3). Mutazioni del GHRH-R nell’uomo sono la causa di rare forme di deficit di GH familiare (4,5).
Un altro ormone secretagogo per il GH è la Ghrelina, prodotta in vari organi oltre che nell’ipotalamo, dove è presente un'elevata espressione del suo recettore (GHSR tipo 1a)(6). Il ruolo di questo secretagogo nella crescita in età evolutiva non è chiaro.
L’ipotalamo del bambino riceve influenze dall’ambiente esterno e da afferenze corticali e sotto-corticali, attraverso i vari neuro-modulatori, neuro-ormoni e neuro-peptidi, come evidente nei casi di c.d. “bassa statura psico-sociale”, dove l’ambiente affettivo del bambino influenza chiaramente la secrezione di GH (il passaggio da un ambiente sfavorevole a uno più favorevole, sia socialmente che affettivamente, porta alla ripresa della crescita staturo-ponderale e a un'aumentata secrezione di GH) (7).
Il GH è una proteina di 191 aminoacidi. L’85-90% del GH circolante ha peso molecolare di 22 kDa; per splicing alternativo si produce una isoforma di 20 kDa che rappresenta il restante 10-15%. Il gene del GH (GH-N) appartiene a una famiglia che comprende i geni per il GH (GH-N, GH-V), per la prolattina (PRL) e per i lattogeni placentari.
La secrezione di GH presenta un modello circadiano con picchi discreti che, in età evolutiva, si verificano soprattutto durante le ore notturne e sono in rapporto con gli stadi del sonno (1,8). La secrezione di GH varia consistentemente con l’età, raggiungendo la massima secrezione durante lo sviluppo puberale e l’adolescenza fino all’età giovane-adulta, per poi diminuire gradualmente con l’avanzare dell’età (9,10).
Il GH in circolo si lega a due proteine vettrici: una a bassa affinità, di significato poco chiaro, che lega preferenzialmente l’isoforma da 20 kDa, e una ad alta affinità (GH-Binding Protein, GHBP) che rappresenta la forma solubile del dominio extra-cellulare del recettore del GH (GHR). Si ritiene che il ruolo della GH-BP sia molteplice: trasporto in circolo del GH, riserva circolante dell’ormone, diminuzione del tasso di degradazione del GH con aumento della sua emivita in circolo.
Il GH esercita i suoi effetti sulla crescita attraverso azioni sul metabolismo e sulla differenziazione cellulare. Molti di questi effetti sono mediati dalla sua attività stimolatoria sulla produzione di fattori di crescita (GFs), e in particolare della IGF-I (Insulin-like Growth Factor–I), prodotta principalmente dal fegato. Lo stimolo alla produzione di IGF-I avviene attraverso il legame del GH al GHR, con l’attivazione di una complessa cascata post-recettoriale. Il legame del GH al GHR porta alla dimerizzazione del recettore stesso sulla superficie cellulare delle cellule bersaglio (fegato, osso, muscolo e vari altri tessuti), con successiva attivazione di diverse vie post-recettoriali. Questi eventi consistono, in sintesi, nell’interazione del GHR con la proteina JAK2, con creazione di siti di ancoraggio per vari mediatori del segnale; reclutamento delle JAK2; reclutamento di proteine STAT, e in particolare della STAT5b; variazioni di processi di fosforilazione e defosforilazione di proteine citoplasmatiche e nucleari; legame di STAT5b al DNA e innesco della trascrizione dei geni bersaglio, in primo luogo per la sintesi di GF. Un’altra via di trasmissione del segnale del GH utilizza il sistema dell’Inositolo Fosfato (IP) e ERK1/2. Difetti dei geni che codificano per ciascuno dei fattori della sequenza post-recettoriale del segnale sono potenziali candidati come cause di diverse forme di bassa statura, e molti di essi sono stati dimostrati dalla letteratura scientifica (11).
I GF sono una serie di proteine, di cui l’IGF-I è quella più direttamente implicata nella crescita staturo-ponderale, in parte GH-dipendente. Il suo ruolo è quello di intervenire nella proliferazione e differenziazione cellulare di vari organi e tessuti e nei processi di crescita, metabolismo, invecchiamento e carcinogenesi. Per quanto riguarda la fase evolutiva, media principalmente l’azione del GH nella crescita delle ossa lunghe attraverso azioni specifiche sulle cartilagini di accrescimento. Circola legata a proteine vettrici (BP), di cui quella a più alta affinità è la IGF-BP3. Come per le GH-BP, il ruolo delle IGF-BP è molteplice: allungamento dell’emivita della IGF-I, funzione di riserva, migliore e più omogenea distribuzione ai vari tessuti e trasporto dell’IGF-I in circolo ai vari bersagli. L’IGF-I, in particolare, circola sottoforma di complesso ternario, formato dalla IGF-I stessa, dalla IGF-BP3 e da una subunità acido-labile (ALS):
- l’IGF-I libera rappresenta circa l’1% dell’IGF-I circolante e ha un’emivita di circa 10 minuti;
- il complesso IGF-I-IGF-BP3 rappresenta il 10-15% dell’IGF-I in circolo e ne porta l’emivita a 30-90 minuti;
- il complesso ternario IGF-I – IGF-BP3 - ALS rappresenta circa l’85% della IGF-I e ne estende l’emivita a più di 12 ore.
L’IGF-I, a sua volta, agisce sulla differenziazione e proliferazione cellulare attraverso l’azione su un recettore (IGFR) che fa parte della famiglia dei recettori per l’insulina. L’attivazione dell’IGFR innesca una cascata post-recettoriale attraverso i sistemi IP e ERK 1/2.
Tutti i diversi step fin qui descritti, dalla sintesi di GHRH all’azione periferica sui tessuti, sono controllati da numerosi geni codificanti e regolatori, la cui alterazione può portare a diverse forme genetiche di patologie della crescita (10,12).
Come tutti gli assi endocrini, anche l’asse GHRH-GH-IGF-I è regolato da meccanismi di feed-back: corto del GH sull’ipotalamo, lungo di IGF-I (e altri fattori influenzati dal GH, come acidi grassi liberi e glucosio) sull’ipotalamo e sull’ipofisi. Il controllo della secrezione di GH comprende quindi due sistemi a circuito chiuso (closed-loop: GH e IGF-I) e uno a circuito aperto (controllo neurale) (6).
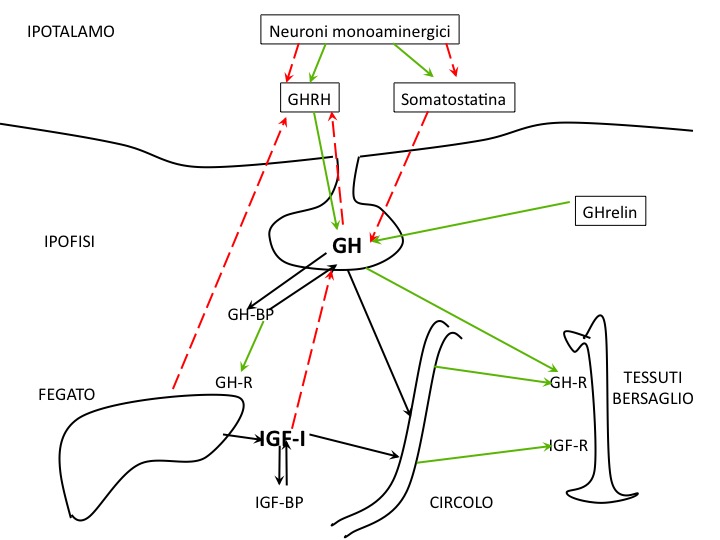
Rappresentazione schematica dell'asse: le frecce verdi rappresentano vie o azioni stimolatorie, quelle rosse inibitorie (vedi testo per le abbreviazioni)
Bibliografia
- Giustina A, Veldhuis JD. Pathophysiology of the neuroregulation of growth hormone secretion in experimental animals and the human. Endocr Rev 1998, 19: 717-97.
- Mayo KE, Cerelli GM, Lebo RV, et al. Gene encoding human growth hormone-releasing factor precursor: structure, sequence, and chromosomal assignment. Proc Natl Acad Sci USA 1985, 82: 63-7.
- Mayo KE, Godfrey PA, Suhr ST, et al. Growth hormone-releasing hormone: synthesis and signaling. Recent Prog Horm Res 1995, 50: 35-73.
- Wajnrajch MP, Gertner JM, Harbison MD, et al. Nonsense mutation in the human growth hormone-releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nat Genet 1996, 12: 88-90.
- Baumann G, Maheshwari H. The Dwarfs of Sindh: severe growth hormone (GH) deficiency caused by a mutation in the GH-releasing hormone receptor gene. Acta Paediatr 1997, S423: 33-8.
- Low MJ. Neuroendocrinology. In: Williams Textbook of Endocrinology 2007.
- Peck MN, Lundberg O. Short stature as an effect of economic and social conditions in childhood. Soc Sci Med 1995, 41: 733-8.
- Buzi F, Zanotti P, Tiberti A, et al. Overnight growth hormone secretion in short children: independence of the sleep pattern.J Clin Endocrinol Metab 1993, 77: 1495-9.
- Hindmarsh PC, Matthews DR, Brook CG. Growth hormone secretion in children determined by time series analysis. Clin Endocrinol (Oxf) 1988, 29: 35-44.
- Dattani M, Preece MA. Growth hormone deficiency and related disorders: insights into causation, diagnosis, and treatment. Lancet 2004, 363: 1977-87.
- Rosenfeld RG, Kofoed E, Little B, et al. Growth hormone insensitivity resulting from post-GH receptor defects. Growth Horm IGF Res 2004, 14 suppl A: S35-8.
- Wit JM, Mullis PE. Genetics of growth. Horm Res Pediatr 2013, 80: 379-80.
Sviluppo staturale: tappe fisiologiche e strumenti di valutazione
Fabio Buzi
SC Pediatria, AO "C. Poma", Mantova
Tappe della crescita
La crescita è un fenomeno complesso, che si effettua attraverso una serie di meccanismi che agiscono sui diversi organi e settori del corpo, secondo tappe che si ripetono nella popolazione in modo regolare. Non consiste solo nell’aumento delle dimensioni del corpo, ma anche nelle modifiche dei rapporti tra diversi segmenti corporei: basti pensare al rapporto tra le dimensioni della testa e quelle del corpo, che nel neonato è di circa 1:4 e nell’adulto diventa circa 1:8.
La crescita nel suo insieme è, fondamentalmente, il risultato dell’interazione tra fattori genetici (es. patrimonio genetico genitoriale, etnia) e ambiente (es. condizioni socio-economiche, nutrizione, componenti culturali in senso lato, epigenetica). Sebbene la crescita non sia un fenomeno continuo e lineare (cioè, analizzata per piccoli intervalli di tempo, si verifica con piccole accelerazioni e decelerazioni, anche con differenze stagionali: ad es. leggermente più accelerata d’estate), può essere rappresentata per intervalli di tempo più lunghi (semestri, anni) come un fenomeno lineare, che, dalla nascita all’età giovane-adulta, si verifica secondo uno schema trifasico:
- fase di rapida crescita staturale e ponderale nei primi (3-4) anni di vita;
- fase di crescita lineare fino all’esordio della pubertà;
- scatto di crescita puberale (spurt) che porta alle dimensioni pressoché definitive e si esaurisce gradualmente fino alla cosiddetta “crescita zero".
Non tutti i settori corporei però seguono questo modello, pertinente più specificamente alla statura (quindi scheletro), ai muscoli e ai principali organi interni: il cervello (e con esso il neuro-cranio) cresce molto rapidamente nei primi 2 anni di vita, raggiungendo a 2-3 anni le dimensioni quasi definitive (come accennato più sopra a proposito del rapporto testa:corpo); il tessuto linfatico raggiunge le massime dimensioni alla pubertà, per poi ridursi fino all’età adulta; l’apparato genitale mantiene invece le dimensioni infantili fino alla pubertà, dove si sviluppa raggiungendo nel giro di pochi anni le dimensioni definitive. Queste differenze sono ben rappresentate nella figura 1, dove le variazioni sono espresse come percentuale raggiunta della dimensione finale dei diversi organi e apparati.
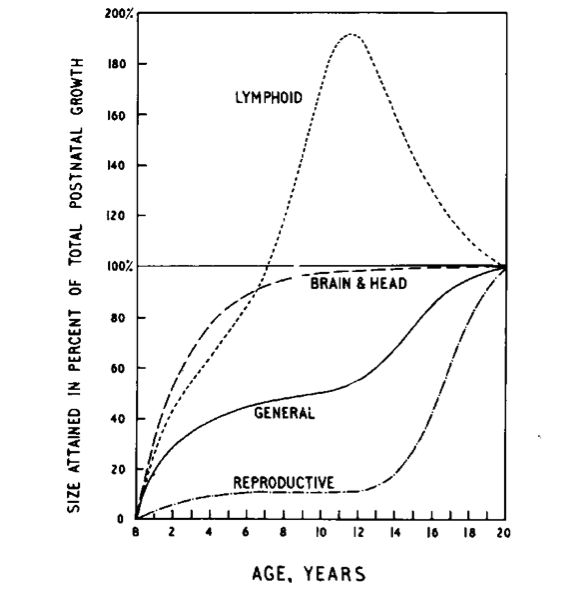
Figura 1 (da 5)
Valutazione della crescita staturale
La crescita staturo-ponderale del bambino è uno dei mezzi più sensibili per valutarne il benessere e la salute. Per poterla studiare correttamente sono però necessari alcuni requisiti:
- l’uso di strumenti precisi;
- l’adozione di una tecnica di misurazione corretta;
- l’utilizzo di standard di crescita adeguati.
L’uso di strumenti precisi si basa sullo stesso principio per il quale si deve usare uno sfigmomanometro ben tarato per misurare la pressione arteriosa o un kit di laboratorio con il minimo di coefficiente di variazione per valutare i livelli di un analita: se così non fosse, rischieremmo di basarci su valori errati per prendere decisioni diagnostiche e terapeutiche. Secondo gli standard internazionali, la misurazione della statura si deve eseguire con strumenti di precisione, come ad es. lo Stadiometro di Harpenden, che fornisce valori approssimati al millimetro, e che rappresenta uno strumento indispensabile in un Centro di Auxologia. La necessità di uno strumento preciso e di una tecnica di misurazione corretta risiede in questo principio: per valutare la velocità di crescita (HV) occorrono almeno due misurazioni a distanza di tempo congrua (normalmente 6 mesi; mai meno di 3 mesi). La HV viene espressa in cm/anno (quindi 12 mesi): è evidente che anche un minimo errore infra- o inter-misuratore verrà moltiplicato per 2 o per 4 se la misurazione viene effettuata su 6 o su 3 mesi, rispettivamente. Quindi un errore di soli 0.5 cm in meno alla prima misurazione e di 0.5 cm in più alla seconda darà un valore in eccesso di 4 cm se la misurazione è stata fatta su 3 mesi o di 2 cm se su 6 mesi, e viceversa!
Piuttosto che utilizzare i classici "statimetri" annessi alla bilancia (piano di appoggio dei piedi mobile, asta centimetrata flessibile, elemento scorrevole sull’asta mobile su perno: altissimo livello di imprecisione dello strumento!), è meglio fissare al muro un centimetro metallico inestensibile a partenza dal piano del pavimento e perfettamente perpendicolare al pavimento stesso e utilizzare poi una squadra per la misurazione del soggetto con tecnica di misurazione corretta.
La tecnica corretta di misurazione è la seguente:
- il soggetto deve stare in piedi (senza scarpe!!) con talloni ravvicinati e adesi al piano posteriore;
- la testa deve essere orientata sul cosiddetto Piano di Francoforte (una linea immaginaria passante per il meato acustico esterno e il margine inferiore dell’orbita deve essere orizzontale);
- il misuratore esercita una lieve pressione verso l’alto sulle mastoidi per favorire la migliore posizione eretta, controllando che il soggetto non alzi i talloni e invitandolo contemporaneamente a inspirare;
- in questo momento leggerà il valore sul contatore dello stadiometro (il piano di misurazione dello stadiometro Harpenden è mobile e controbilanciato, e il contatore è a fianco dello stesso) o, in caso di attrezzo più “artigianale”, sul centimetro metallico millimetrato fissato al muro.
Questa modalità è impossibile per bambini piccoli che non mantengono la posizione eretta o che si oppongono alla misurazione, ciò che avviene praticamente sempre sotto i 2 anni di età e spesso in bambini di giovane età (fino a 3-4 anni) piuttosto oppositivi. Esistono a questo proposito stadiometri orizzontali (Infantometro, Stadiometro orizzontale), che necessitano della presenza di due operatori per la misurazione corretta del soggetto, con tecnica analoga a quella già descritta.
La tipica curva di crescita è rappresentata in figura 2, dove si possono distinguere le 3 fasi principali descritte precedentemente:
- una prima fase di crescita rapida dalla nascita fino ai 3-4 anni, per cui la lunghezza alla nascita aumenta del 50% circa alla fine del primo anno e del 60% alla fine del secondo, con pendenza elevata che gradualmente diminuisce fino ai 4 anni circa;
- una seconda fase piuttosto regolare fino all’inizio della fase puberale;
- una terza fase di crescita rapida (scatto di crescita puberale, spurt) e successivo arresto accrescitivo, corrispondenti alla pubertà e al raggiungimento della statura definitiva.
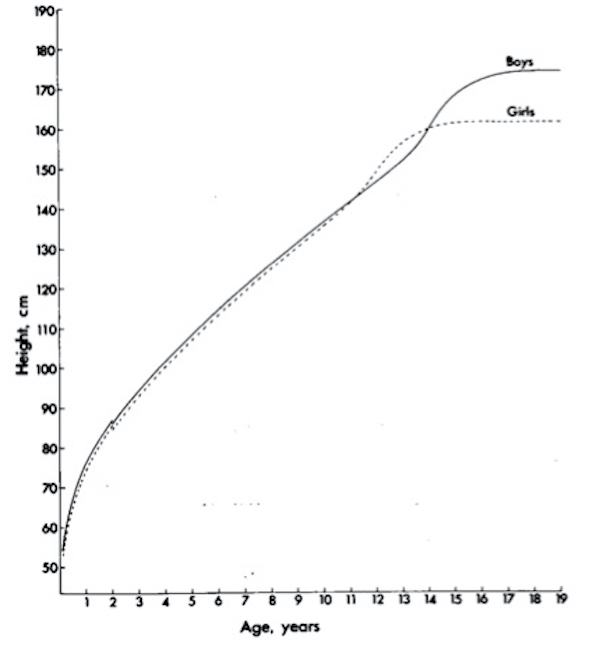
Figura 2. Evoluzione della statura rispetto all'età nei 2 sessi (mod da 6)
Nella prima e nella seconda fase le differenze statistiche tra maschi e femmine sono trascurabili, mentre nella terza fase si stabiliscono le differenze staturali tipiche tra i due sessi. Durante la prima e la seconda fase il maschio “medio” (cioè di peso medio alla nascita e che cresce a velocità media lungo il 50° percentile) ha una statura minimamente superiore alla femmina “media”. La femmina entra però in pubertà mediamente prima del maschio e presenta lo spurt circa 2 anni prima di quest’ultimo, superandolo temporaneamente in altezza. Il maschio successivamente la risupera dopo lo spurt e raggiunge un’altezza finale di circa 12-13 cm maggiore: questa differenza è costante per maschi e femmine adulti appartenenti allo stesso percentile, ed è uno dei presupposti per il calcolo dell’altezza media dei genitori corretta per sesso (v. oltre).
Velocità di crescita
Mentre questo modello generale di crescita è valido per tutti i bambini, esiste una grande variabilità individuale, sia per quanto riguarda l’entità dell’altezza sia per i tempi in cui viene iniziata e conclusa la fase puberale (soggetti di altezza più bassa, media e più alta; soggetti che maturano più lentamente o più rapidamente). Il modello di crescita appena descritto viene chiamato curva “di distanza”, perchè rappresenta la distanza in termini di altezza percorsa dal bambino: in qualche modo è come se dovessimo misurare la distanza tra due città in termini di km. Questo modello non ci dice però in quanto tempo e in che modo (accelerazioni, decelerazioni) questa distanza viene coperta. Lo si può quindi trasformare in curva di velocità, mantenendo sull’asse delle x l’età cronologica e sostituendo all’altezza sull’asse delle y la velocità di crescita (Height Velocity, HV) in cm/anno: è come se mettessimo i km/ora di un mezzo che percorre la distanza tra le due città di cui sopra (figura 3). In questo caso tutti i fenomeni precedentemente descritti vengono amplificati, e si può cogliere più facilmente la fase di alta velocità dei primi 2-3 anni di vita, la fase di crescita regolare successiva e la 3° fase puberale con lo spurt e il successivo decremento fino alla crescita zero. Questo modello è certamente più sensibile nel cogliere eventuali variazioni individuali anche minime nel ritmo di crescita, ed è quindi clinicamente importante. La velocità di crescita si esprime in cm/anno, quindi per calcolarla servirebbe misurare la statura in 2 successive rilevazioni a distanza di 12 mesi. Poiché questo non è sempre pratico (o desiderabile), si utilizzano comunque 2 rilevazioni (ad almeno 3 mesi di distanza, meglio 6), normalizzate a 12 mesi con una semplice proporzione:
velocità (cm/anno) = delta statura/frazione di tempo
dove delta statura è la differenza in cm tra le due misurazioni in quell'intervallo di tempo e frazione di tempo è l'intervallo fra le 2 misurazioni calcolato in decimali di anno (esempio: 3 mesi = 3/12 = 0.25; 6 mesi = 6/12 = 0.5, ecc.). Esiste a questo proposito una tabella dei millesimi di anno che si impara ad usare facilmente ed è più precisa; una scorciatoia può essere dividere il numero di giorni dell'intervallo per 365: ad esempio se l'intervallo tra le due misurazioni fosse 95 giorni (95/365 = 0.26) e in quell'intervallo il paziente fosse cresciuto di 1.8 cm, la sua velocità in cm/anno sarebbe 1.8/0.26 = 6.9 cm/anno.
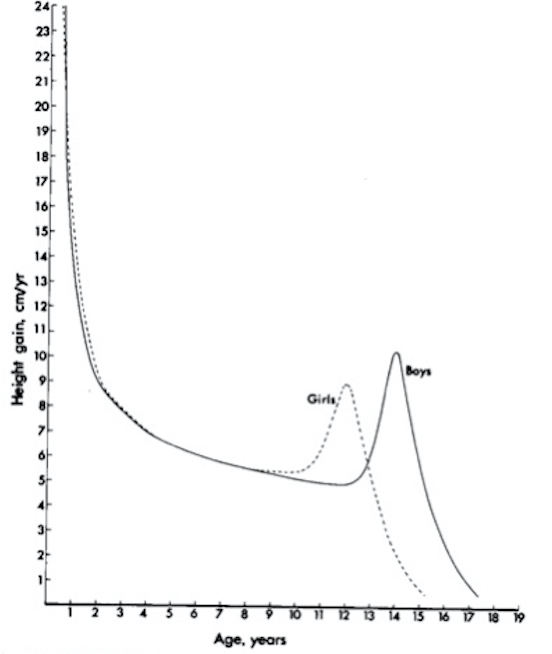
Figura 3. Rappresentazione grafica della velocità di crescita alle diverse età nei 2 sessi (da 6)
I due modelli descritti (curva di “distanza” e curva di “velocità”) costituiscono i presupposti per le tabelle di crescita normalmente conosciute come curve dei percentili o degli Standard di crescita. Questi consistono in tabelle o grafici che permettono di confrontare l’altezza o la HV di un soggetto in esame con quelle di una popolazione di riferimento. Si basano sulla raccolta di dati di un ampio campione rappresentativo della popolazione di riferimento. Normalmente sono rappresentati con linee che identificano diversi percentili (o centili); 3° (o 5°), 10°, 25°, 50°, 75°, 90° e 97° (o 95°), comprendendo quindi la distribuzione al 94% (o 90%) di una determinata variabile della popolazione (altezza, HV, ma anche circonferenza cranica, segmenti corporei, ecc.). La scelta di utilizzare come estremi della distribuzione il 3° e il 97° piuttosto che il 5° e il 95° percentile è del tutto arbitraria e non rappresenta altro che il 94% delle osservazioni nel primo caso e il 90% nel secondo. Poiché l’altezza (come pure la HV e altre variabili antropometriche: statura da seduti, lunghezza arti inferiori, diametro bi-trocanterico, diametro bis-acromiale, circonferenza cranica, ecc.) si distribuisce in modo Gaussiano, può essere espressa anche in termini di deviazione standard (SD) dalla media, in quanto in tal caso la mediana (50° percentile della distribuzione) corrisponde praticamente alla media aritmetica della distribuzione. In questo senso:
- +1 e -1 SD corrispondono rispettivamente all’84° e al 16° centile e comprendono il 78% delle osservazioni;
- +2 e -2 SD corrispondono rispettivamente al 98° e al 2° centile e comprendono il 96% delle osservazioni
- +3 e -3 SD comprendono praticamente tutte le osservazioni (99.8%).
In particolare:
- il 75° e il 25° centile corrispondono rispettivamente a +0.64 e -0.64 SD;
- il 90° e il 10° centile corrispondono rispettivamente a +1.28 e -1.28 SD;
- il 97° e il 3° centile corrispondono rispettivamente a +1.88 e -1.88 SD;
- il 50° centile corrisponde ovviamente a 0 SD.
Poiché, però, i bambini crescono e i valori di altezza e delle altre variabili antropometriche cambiano con l’età e con il genere, è necessario rappresentare tali valori in termini di SD come punteggi standardizzati per l’età e per il genere, utilizzando le medie e le SD pubblicate per le diverse popolazioni e per le diverse variabili e trasformandole in Standard Deviation Score (SDS: punteggi standardizzati, appunto), attraverso il seguente calcolo:
SDS = (x-X)/SD
dove x è la misura rilevata nel soggetto in esame, X la media di tale variabile calcolata per età e genere nella popolazione di riferimento, e SD la SD calcolata per la stessa età e genere. Ciò significa, per esempio, che una bimba di 4 anni con statura sul 25° centile, in base alle equivalenze sopra riportate, avrà una SDS di –0.64. Questo vale anche per la HV, ma con diverso criterio. A differenza della curva di distanza, infatti, la curva di velocità deve essere interpretata in un modo diverso: qualunque sia il centile di distanza sul quale il bambino cresce, il modello di crescita è sempre lo stesso. Come si può osservare dalle curve dei percentili (figura 4), i “canali” di crescita (3°-10°, 10°-25° centile, ecc.) sono praticamente paralleli tra loro: ne deriva che un soggetto che cresce sul 10° percentile di distanza dovrà avere la stessa velocità di uno che cresca sul 75° o sul 50°: questa velocità dovrà essere intorno al 50° centile e variare dal 25° al 75° nell’arco di uno o più anni. Un soggetto cioè che cresca costantemente (per più anni) anche poco al di sotto del 25° percentile di HV “taglierà” i centili di distanza verso il basso, “perdendo terreno”; il contrario per uno che cresca sempre al di sopra del 75°.
Anche la HV può essere espressa come SDS, con l’attenzione di riferire l’età al punto di mezzo tra due successive misurazioni, in quanto la HV è uno spazio/tempo e quindi può essere rappresentata non da un punto ma da un segmento, i cui estremi sono l’età della prima misurazione e quella della misurazione successiva. Ad esempio, per un bambino che venga misurato la prima volta a 6 anni e la seconda volta a 6 anni e mezzo, l’età a cui riferire il calcolo della SDS di HV (HV-SDS) sarà 6 anni e 3 mesi.
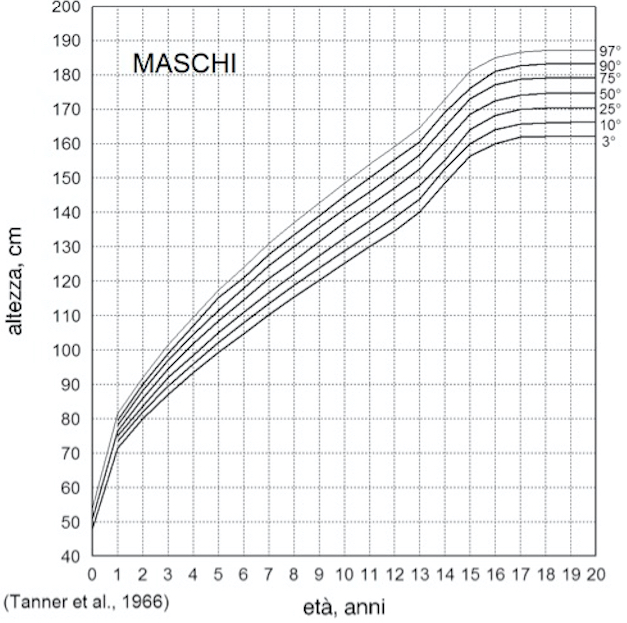
Figura 4. Percentili di crescita. Dopo aver misurato la statura del bambino, la rappresento con un punto all'incrocio fra l'età in ascissa e l'altezza in ordinata. Eseguendo misurazioni seriate e unendo con una linea i relativi punti, posso identificare se la crescita è regolare (segue il suo percentile), oppure tende a tagliare i percentili verso l'alto (crescita eccessiva) o verso il basso (deficit di crescita)
Standard di riferimento
La scelta degli standard da usare per altezza e HV è un altro argomento spesso dibattuto. Le carte di crescita più “complete”, elaborate con una metodologia di campionamento molto precisa, sono quelle di Tanner e Whitehouse e Tanner e Davies (popolazione inglese e popolazione statunitense rispettivamente). Sono certamente le più diffuse, ma non sono rappresentative della nostra popolazione, sia per l’origine che per la cronologia del campionamento (Inghilterra anni ’60 e successivi aggiornamenti, USA anni ’80). Inoltre, presuppongono la trasformazione dell’età “anni + mesi” in età decimale (anno + decimale di anno), calcolo molto semplice ma poco utilizzato perché implica l’uso della tabella dei millesimi di anno di Tanner. Esistono ormai da anni curve di crescita staturale e ponderale rappresentative della realtà italiana dai 2 ai 19 anni (3) (figura 5). Va comunque osservato che la popolazione è sempre più composita, dati i flussi migratori e gli standard dovrebbero quindi essere continuamente aggiornati. Le carte di crescita devono in ogni caso essere considerate degli strumenti da utilizzare criticamente, tenendo conto di altre componenti della crescita, come i condizionamenti genetici (es. stature dei genitori, etnia del soggetto in esame), ambientali (nutrizione, eccesso o difetto ponderale, ecc.) e maturazione biologica (età ossea).
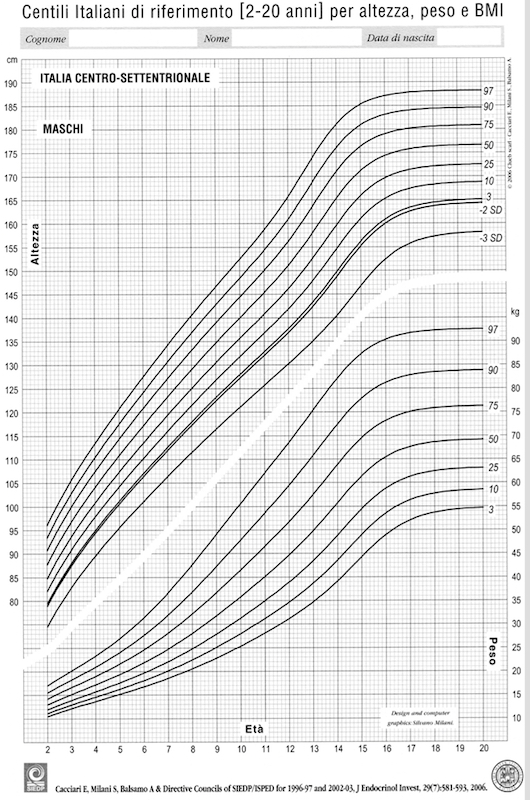
Figura 5. Curve di crescita staturale e ponderale per i maschi italiani (da 3)
Qualunque sia la curva standard di crescita utilizzata, bisogna infine ricordare che la variabile più importante è la HV. A questo proposito, nonostante siano state elaborate diverse carte di crescita di distanza per statura e peso, le curve di HV tuttora universalmente utilizzate per la HV e per il calcolo della HV-SDS sono quelle di Tanner e Whitehouse (figura 6), in quanto costruite con una metodologia di campionamento molto precisa e specifica (longitudinale) e non derivate semplicemente dalla rielaborazione dei dati (trasversali) di crescita lineare. Per le età da 0 a 2 anni esistono poi carte più dettagliate, che prendono in considerazione la lunghezza supina (0-2 anni) e quella eretta fino ai 5 anni e mezzo (Tanner 1973) o quelle della NCHS o della WHO e molte altre ancora. Anche in questo caso, come già detto, è bene decidere di utilizzare un certo tipo di standard cui abituarsi e usarlo con le attenzioni già esposte precedentemente.
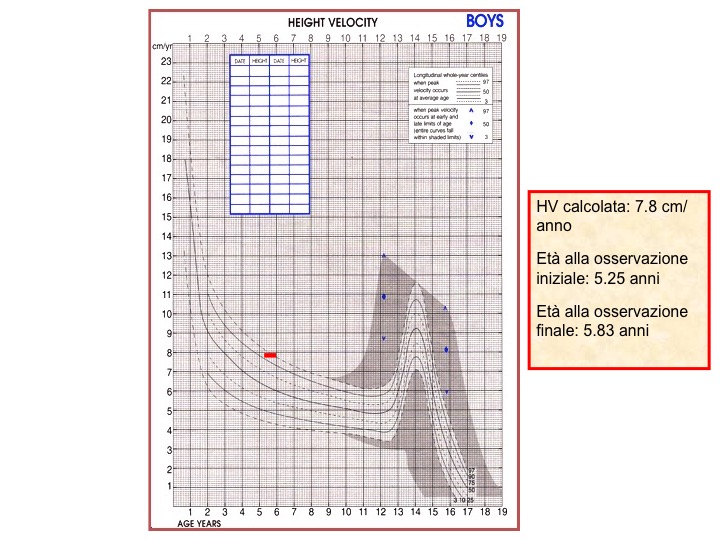
Figura 6. Curva della velocità di crescita. la HV è una velocità, quindi un "spazio su tempo", e si svolge quindi nel corso del tempo e non in un piccolo istante (cioè al momento della visita, come per la rilevazione dell'altezza o del peso). La HV sarà quindi rappresentata da un segmento (e non da un punto, come per l'altezza o il peso), che avrà come estremi l'età della prima rilevazione e l'età della seconda rilevazione; il percentile sarà riferito al punto di mezzo di tale segmento. Il bambino dell'esempio sopra ha una velocità di crescita che lo colloca tra il 90° e il 97° percentile
Bersaglio genetico
La crescita è fortemente condizionata geneticamente, in particolare dalle stature dei genitori. È possibile, quindi, valutare l’altezza di un soggetto in rapporto a quelle dei suoi genitori. Un metodo pratico è quello della costruzione del “bersaglio genetico”, che si basa sulla già citata differenza statistica di circa 12.5 cm (12–13 cm) tra altezza media dei maschi e altezza media delle femmine da adulti. È quindi possibile trasformare il percentile di altezza di un padre nel percentile femminile sottraendo 12.5 cm alla sua altezza reale e allo stesso modo trasformare quella di una madre in percentile maschile addizionando alla sua altezza i 12.5 cm. Già in questo modo si potrà vedere se la statura del soggetto in esame, maschio o femmina che sia, si trovi nella stessa posizione occupata da uno o entrambi i genitori. La costruzione del “bersaglio genetico” consiste poi nel calcolo della statura media dei genitori corretta per genere (Mid Parental Height, MPH), secondo il modello (tutte le misure sono in cm):
- MPH maschio = (altezza paterna + altezza materna + 12.5)/2;
- MPH femmina = (altezza paterna + altezza materna - 12.5)/2.
È possibile calcolare la SDS della MPH (MPH-SDS) rapportando il valore della MPH al valore della statura finale di una femmina o di un maschio (età: 19 anni), e confrontare così la altezza-SDS del soggetto in esame (femmina o maschio) con la MPH-SDS dei suoi genitori.
Infine, si può calcolare il “bersaglio genetico” aggiungendo alla MPH ± 10 cm per i maschi e ± 9 cm per le femmine: questi limiti ci danno la MPH ±2 SD per quella coppia genitoriale, in quanto 1 DS per le stature da adulti è 5 cm per i maschi e 4.5 per le femmine. Questo calcolo valuta la statura attesa del 96% dei figli di un determinato sesso di quella coppia genitoriale e non è una previsione di statura finale del singolo soggetto in esame! Si può essere più "conservativi" nel definire il bersaglio, restringendone i limiti: ad esempio +8 e -8 cm dalla MPH o ancora meno.
Crescita ponderale
Il peso deve essere rilevato per mezzo di una bilancia ben tarata (bilancia pesa-infanti per le età da 0 a 2-3 anni e bilancia a stadera o elettronica per le età successive), che assicuri un’approssimazione a 0.1 kg. Il soggetto deve essere pesato nudo, meglio se al mattino a digiuno e dopo aver evacuato. La crescita ponderale non è molto diversa da quella staturale e presenta le stesse fasi e le stesse variazioni inter-individuali. Esistono anche per il peso le carte di crescita rappresentate in centili. Essendo il peso una variabile non distribuita in modo Gaussiano (poiché risente fortemente dei condizionamenti ambientali e nutrizionali), non è corretta la sua rappresentazione in termini di SDS: è possibile però calcolare per le diverse età l’Indice di Massa Corporea (Body Mass Index, BMI) e rapportarlo alle carte specifiche pubblicate (in Italia sono disponibili quelle di Cacciari et al).
Valutazione delle proporzioni corporee
Esistono inoltre metodi per valutare le proporzioni corporee dei bambini durante la crescita e individuare possibili asimmetrie e disarmonie.
Un sistema è quello della rilevazione del rapporto segmento superiore/segmento inferiore. Un metodo è quello di confrontare la statura da seduto (Sitting Height, SH) con la lunghezza sub-ischiatica (Sub-ischial Leg Length, SLL) per mezzo di uno strumento specifico derivato dallo stadiometro di Harpenden, applicato a uno sgabello indeformabile di lunghezza fissa o semplicemente utilizzando un analogo sgabello appoggiato allo stadiometro:
- SH = altezza misurata con lo stadiometro - altezza dello sgabello;
- SLL = altezza del soggetto - SH.
Anche per queste variabili esistono carte dei percentili specifiche.
Un altro metodo più semplice è quello della rilevazione dello span (apertura estrema delle braccia e sua misurazione) e confronto con l’altezza del soggetto: normalmente lo span non eccede di ± 5 cm l’altezza.
Variazioni più specifiche delle proporzioni corporee vengono poi ottenute per mezzo dell’antropometro, uno strumento modulare finalizzato alla misurazione di singoli segmenti corporei (lunghezza degli arti, diametri vari – cranico, bis-acromiale, bi-trocanterico, ecc.) di pertinenza però decisamente specialistica (es. dismorfologia, genetica clinica).
Bibliografia
- Benso L, Conrieri M. Metodiche di valutazione auxologica. In: Quaderni di endocrinologia pediatrica. G. Tonini Ed. Vol. I, Archimedica, Torino 1996.
- Buzi F. Gli standard di crescita: le curve di riferimento. Attendibilità e utilizzo pratico. In: Quaderni di endocrinologia pediatrica. G. Tonini Ed. Vol. I, Archimedica, Torino 1996.
- Cacciari E, Milani S, Balsamo A, et al. Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocrinol Invest 2006, 29: 581-93.
- Nicoletti I. Auxologia normale e patologica. Ed. Centro Studi Auxologici. Firenze, 1994.
- Tanner JM. Foetus into man. 2nd Ed, 1989. Castelmead Publications, Ware, UK.
- Tanner JM, Whitehouse RH, Takahishi M. Standards from birth to maturity for height, weight, height velocity, and weight velocity: British children 1965. Arch Dis Child 1966, 41: 454-71 e 613-35.
- Tanner JM, Whitehouse RH. Clinical longitudinal standards for height, weight, height velocity, weight velocity, and stages of puberty. Arch Dis Child 1976, 51: 170-9.
- Tanner JM, Davies PSW. Clinical longitudinal standards for height and height velocity for North American Children. J Pediatr 1985, 107: 317-29.
Valutazione dell'età ossea
Fabio Buzi
SC Pediatria, AO "C. Poma", Mantova
I bambini non crescono tutti allo stesso ritmo e non maturano tutti allo stesso modo e nello stesso tempo. Questo è molto evidente all’età della pubertà, quando troviamo soggetti già fisicamente “adulti” e coetanei ancora totalmente infantili: basti pensare ai componenti, maschili e femminili, di una seconda o terza media. Queste differenze sono comunque già presenti a tutte le età, fin dalla nascita. Per esempio, le femmine sono, in media, biologicamente più avanti rispetto ai maschi già dalla metà della vita fetale in poi. Allo stesso modo, alcuni bambini (maschi e femmine) sono più precoci nella maturazione rispetto ad altri nel corso della fase evolutiva pre- e post-natale: per citare J.M. Tanner, pioniere dell’Auxologia, “l’analogia ci viene dalla musica classica: alcuni bambini suonano la loro crescita come “andante”, altri come “allegro”, e un numero minore come “lentissimo”. Sembra che l’eredità giochi un grande ruolo nell’impostare il metronomo, ma noi non ne conosciamo il meccanismo fisiologico”.
È evidente come sia necessaria una misura della maturazione biologica di un bambino, per distinguere nell’ambito della fisiologia i “maturatori normali” dai “tardi maturatori” e dai “maturatori precoci”, e cogliere allo stesso modo i ritardi o gli anticipi patologici. Tra i possibili indicatori di maturazione biologica, l’età staturale, l’età ponderale, l’età psicologica e l’età dentale sono assolutamente inadeguate a questo compito, perché risentono di troppe influenze ambientali, nutrizionali, genetiche, culturali e sociali. L’indicatore più affidabile di maturazione biologica è l’età ossea.
La maturità scheletrica è una buona misura di quanto le ossa di un certo settore siano progredite nel processo di maturazione, non tanto in grandezza quanto nella loro forma e nella loro posizione rispetto alle altre ossa contigue, e questo è possibile valutarlo per mezzo di una semplice radiografia. Ogni segmento osseo inizia la sua maturazione prima come centro di ossificazione primario, per poi passare attraverso diversi stadi di ingrandimento e modellamento fino alla forma definitiva: molte ossa, come le ossa lunghe, presentano più centri di maturazione (es. epifisi), che maturano indipendentemente dal centro di maturazione principale, e alla fine raggiungono la forma finale attraverso la fusione delle epifisi con la metafisi dell’osso stesso. Questo è ben evidenziabile con una radiografia diretta dell’osso, in cui i centri di ossificazione assumono aspetti radiologici diversi durante le varie fasi maturative, aspetti morfologici che possono essere utilizzati come indicatori discriminanti delle singole tappe maturative. Sebbene queste modificazioni costituiscano un processo continuo e non delle vere tappe distinte, la loro valutazione può essere ridotta a indicatori di sviluppo discreti.
Teoricamente qualsiasi segmento osseo corporeo potrebbe essere utilizzato a questo fine, ma in pratica le ossa della mano e del polso costituiscono la parte più adatta alla valutazione della maturità scheletrica, per l’elevato numero di ossa corte (carpo) e ossa lunghe (falangi), e per la semplicità di esecuzione di una radiografia diretta della mano. Gli “indicatori di maturità” (cioè i segmenti ossei – nuclei di ossificazione - delle ossa di radio, ulna, carpo, ossa metacarpali e falangi) sono pressoché gli stessi nei due principali metodi impiegati al mondo per la determinazione dell’età ossea: il metodo di Greulich e Pyle (G&P) (successive edizioni: 1950 e 1959) e quello di Tanner e Whitehouse (TW), nelle sue successive edizioni (TW1, TW2 e TW3). Questi due metodi sono stati resi disponibili sottoforma di atlanti da utilizzare nella pratica clinica. Viene esplicitato nell’atlante il metodo di esecuzione della radiografia (posizione della mano, distanza dalla fonte di raggi, ecc). Per convenzione gli indicatori sono stati studiati sulla mano sinistra di un campione di soggetti, femmine e maschi separatamente, in diverse fasi evolutive, in modo longitudinale (sempre i medesimi soggetti durante la loro crescita). La scelta della mano sinistra è dovuta al fatto che, al tempo del campionamento (G&P: anni 1920-50, USA; TW: 1950–70, UK) la mano sinistra era quella meno frequentemente menomata (a quel tempo ancora molti ragazzi lavoravano nelle fabbriche e potevano aver subito incidenti sul lavoro): non ha senso quindi utilizzare la mano destra nei mancini come avviene spesso in alcune Radiologie, perché gli standard di riferimento sono stati rilevati comunque sulla mano sinistra.
In pratica ciascun metodo dovrebbe essere utilizzato seguendo attentamente le istruzioni in esso contenute: in realtà questo non sempre avviene, specie per il metodo G&P, in cui sono presentate radiografie campione a successive età dell’intera mano e polso separatamente per femmine e maschi, per cui la valutazione avviene spesso affrettatamente per semplice confronto tra la radiografia del soggetto in esame e quella dell’atlante più simile dal punto di vista morfologico. Questo può in realtà portare a errori di valutazione.
Esaminiamo brevemente i due metodi più usati.
Nell’atlante G&P (figura 1) il metodo corretto di valutazione, suggerito dagli Autori, sarebbe il seguente:
- confrontare la radiografia del soggetto in esame con lo standard dello stesso genere e dell’età cronologica più vicina;
- confrontare la radiografia con gli standard adiacenti;
- scegliere lo standard che sembra assomigliare più fedelmente alla radiografia in esame;
- esaminare ciascun segmento osseo in sequenza ordinata, attribuendo ai singoli segmenti l’età ossea corrispondente, secondo le istruzioni contenute nel testo dell’atlante;
- infine, arrivati allo standard più somigliante, attribuire l’età ossea corrispondente o estrapolarne una intermedia tra le due più simili.
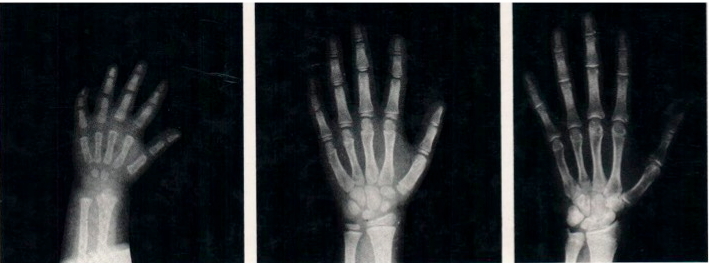
Figura 1. Metodo a confronto (es. Greulich& Pyle): la radiografia del paziente viene confrontata con immagini campione in sequenza temporale evolutiva pubblicate su un Atlante di riferimento; al-alla paziente in esame viene attribuita l’età ossea dell’immagine di riferimento che più si avvicina (per caratteristiche morfologiche e maturative delle ossa del polso e delle falangi) alla Rx in esame; nell’Atlante di Greulich& Pyle, ad esempio, le radiografie campione sono divise per sesso e sono in progressione di 6 mesi dalla nascita alla maturità.
Questa procedura è stata poi perfezionata ulteriormente da Roche rendendola più rigida, ma con tempi di lettura piuttosto lunghi.
Con il metodo TW (figure 2 e 3) la scelta di presentare le immagini dei singoli segmenti ossei e non della mano e polso per intero è stata finalizzata a evitare eccessiva soggettività e letture frettolose “per confronto diretto”. Consiste nell’attribuzione di un punteggio a 20 segmenti ossei scelti tra i nuclei di ossificazione delle epifisi distali di radio e ulna e di quelli delle ossa metacarpali e delle falangi (13 segmenti) più 7 ossa del carpo. Il sistema è un po’ più complesso rispetto a quello di G&P (che, forse anche per questa ragione, è quello comunque più usato a livello mondiale) e implica una fase di addestramento dell’operatore, che porta alla pressoché completa memorizzazione dei criteri di punteggio, accelerando così i tempi di esecuzione della lettura. Devono essere rigorosamente seguite le indicazioni del testo sull’attribuzione di uno stadio maturativo rispetto al precedente o successivo, pena risultati del tutto inaffidabili. Per facilitare l’operatore, gli indicatori di maturità di ciascun osso e di ciascuno stadio sono stati esemplificati graficamente accanto alle immagini radiologiche del segmento in esame. La somma dei punteggi attribuiti a ciascun segmento conduce all’età ossea del soggetto in esame. Mentre nella versione TW1 il punteggio derivava dalla valutazione di tutte le 20 ossa selezionate dal metodo, nell’aggiornamento TW2 vengono distinte 3 diverse modalità: punteggio “20 ossa” (come nel TW1), punteggio “RUS” (radio, ulna e ossa metacarpali e falangee) (quest'ultimo più utile nella valutazione clinica – endocrinologica) e “CARPAL”, limitato alle sole ossa del carpo. La successiva versione (TW3) prende invece in considerazione le sole ossa RUS ed è fornita con software per l'utilizzo tramite computer.
Figura 2. Segmenti ossei presi in considerazione nel metodo a punteggio di Tanner& Whitehouse (TW). Le versioni più utilizzate sono 2:
-
TW2, a sua volta suddivisa in 3 sottotipi:
-
«20 ossa», che considera tutti i 20 segmenti scelti, sia del carpo che di radio, ulna, metacarpo e falangi;
-
«RUS» (Radio Ulna Short bones), che utilizza solo radio, ulna, metacarpo e falangi;
-
Carpo, che utilizza solo i segmenti ossei del carpo;
-
-
TW3, la più recente, che utilizza solo le ossa «RUS».
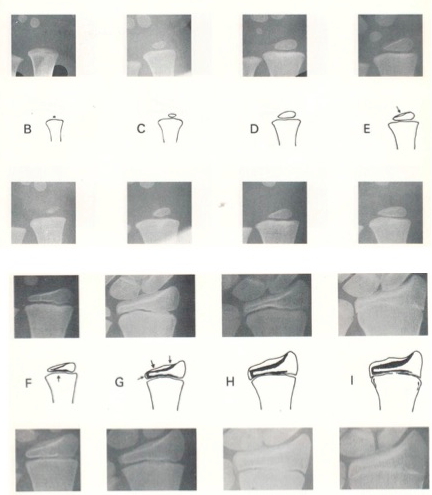
Figura 3. Esempio di metodo a punteggio (Tanner Whitehouse: TW): i diversi segmenti ossei presi in considerazione sono schematizzati con disegni che esemplificano altrettanti criteri morfologici per l’attribuzione di un punteggio (B,C,D,E,F,G,H,I) con l’aggiunta di immagini radiografiche che aiutano l’operatore nella scelta dello stadio da attribuire: la somma dei punteggi dei singoli segmenti (epifisi distale di radio e ulna, nuclei di ossificazione del I, III e V metacarpo, delle falangi prossimali di I, III e V dito, delle falangi medie di III e V dito, e delle falangi distali di I, III e V dito, presi in considerazione nell’ultima versione del metodo TW, chiamata «TW3») corrisponde alla «età ossea» del soggetto in esame. Per applicare questo metodo (TW, nelle versioni TW2 e TW3) è necessario un «training» specifico teorico e pratico. Nella figura è riportata la schematizzazione per l’epifisi distale del radio (modificato da Tanner VJM, Whitehouse RH, Cameron N. Valutazione della maturazione scheletrica e predizione dell'altezza adulta. Metodo TW2. Ed. italiana a cura di A. Tenore. Martinucci Pubblicazioni Mediche, Napoli, 1990).
Con entrambi i metodi può inoltre essere attuata una previsione della statura finale più probabile: per il metodo G&P con il sistema di Bailey e Pinneau; con i metodi TW con il sistema Tanner-Whitehouse. Le equazioni di predizione prendono in considerazione l’età cronologica e il genere del soggetto e la sua età ossea; può essere aggiunta, se disponibile, la velocità di crescita e, sempre se disponibile, la differenza in età ossea a distanza di un anno. Questa previsione viene poi utilizzata confrontandola con il “bersaglio genetico” nella valutazione complessiva della crescita del soggetto in esame.
Esistono ancora ulteriori metodi di lettura dell’età ossea, come il metodo FELS, che però è molto complesso e non utilizzato correntemente nella pratica clinica, così come modifiche dei metodi sopra esposti apportate da singole scuole auxologiche.
Il consiglio è quello di scegliere un metodo di lettura e utilizzarlo correntemente, possibilmente affidandolo a uno o due valutatori, sempre gli stessi per ciascun Centro, che divengano esperti e quindi il più possibile affidabili, ricordando che l’età ossea non è un parametro “certo”, ma che contribuisce, assieme alle altre variabili auxologiche e cliniche, a formulare un giudizio complessivo sulla crescita del soggetto in esame.
Bibliografia
- Aicardi G, Vignolo M, Di Battista E, Mattei R. La valutazione dell’età scheletrica. In: Quaderni di endocrinologia pediatrica. G. Tonini Ed. Vol. I, Archimedica, Torino 1996.
- Bailey N, Pinneau SR. Tables for predicting adult height from skeletal age: revised for use with the Greulich-Pyle hand standards. J Pediatr 1952, 40: 423-41.
- Greulich WW, Pyle SI. Radiographic atlas of skeletal development of the hand and wrist. 2nd Ed, Stanford University Press, Stanford, California, 1959.
- Roche AF. The measurement of skeletal maturation. In: Johnston FE, Roche AF, Susanne C, Eds. Human physical growth and maturation. Plenum Press, New York 1979.
- Tanner JM. Foetus into man. 2nd Ed, Castlemead Publications, Ware, UK, 1989.
- Tanner JM, Whitehouse RH, Marshall WA, et al. Assessment of skeletal maturity and prediction of adult height. 2nd Ed, Academic Press, London, 1983.
- Tanner JM, Healy M, Goldstein H, et al. Assessment of skeletal maturity and prediction of adult height (TW3 Method), 3rd Ed, WB Saunders, Harcourt Publishers Ltd, London, 2001.
- Vignolo M, Milani S, Cerbello G, et al. FELS, Greulich-Pyle, and Tanner-Whitehouse bone age assessments in a group of Italian children and adolescents. Am J Hum Biol 1992, 4: 493-500.
Tappe fisiologiche e strumenti di valutazione dello sviluppo puberale maschile
Gianni Russo & Alessandra di Lascio
Centro di endocrinologia dell’infanzia e dell’adolescenza, Ospedale San Raffaele, Milano
La pubertà è il periodo di transizione tra l’infanzia e l’età adulta durante il quale si realizzano cambiamenti ormonali, somatici, psicologici e metabolici, che portano l’individuo a completare lo sviluppo dei caratteri sessuali secondari, acquisire capacità riproduttiva e stabilire un’identità sessuale di genere.
I principali cambiamenti che avvengono durante la pubertà sono:
- sviluppo dei caratteri sessuali primari (gonadi) e secondari, genitali ed extra-genitali (peluria pubica e ascellare, barba e baffi);
- rapido incremento staturale e modificazioni della composizione corporea;
- raggiungimento della fertilità;
- stabilirsi dell’identità di genere.
Vi è un’ampia variabilità cronologica della pubertà fisiologica, che normalmente va per i maschi dai 9 ½ ai 13 ½ anni. Tale variabilità è legata a fattori ambientali, etnici e socio-economici. La pubertà si completa nell’arco di 3-5 anni.
Asse ipotalamo-ipofisi-gonadi (HPG)
Durante la pubertà si realizzano diverse variazioni ormonali, che comprendono attivazione dell’asse HPG, aumento di steroidi surrenalici e aumento della produzione di GH.
Il principale meccanismo che induce la maturazione puberale è l’attivazione dell’asse HPG. L’ipotalamo e l’ipofisi sono due strutture, strettamente collegate tra loro, situate alla base del cranio che collegano il sistema nervoso centrale (SNC) e il sistema endocrino. L’ipotalamo è il centro da cui partono gli impulsi che regolano il ritmo sonno/veglia, la fame, la sete, la temperatura corporea e il sistema endocrino. Esso produce sostanze, dette neuro-ormoni, che stimolano l’ipofisi a produrre gli ormoni ipofisari. Tra le sostanze prodotte dall’ipotalamo c’è l’ormone stimolante la secrezione di gonadotropine (GnRH), che a partire dall’epoca puberale viene secreto in maniera pulsatile.
La produzione ipotalamica di GnRH è presente anche durante l’epoca fetale e la prima infanzia: tale fenomeno è noto come minipuberty, in quanto i valori di gonadotropine, testosterone e inibina risultano aumentati. Solitamente, dopo i primi 6 mesi di vita la secrezione di GnRH si silenzia fino all’epoca puberale.
Il GnRH agisce su specifici recettori localizzati sulla membrana plasmatica delle cellule gonadotrope dell’adeno-ipofisi, stimolando la produzione delle gonadotropine ipofisarie, LH e FSH. Le gonadotropine, secrete in modo pulsatile, a loro volta agiscono perifericamente su specifici recettori dei testicoli, stimolando la produzione di androgeni da parte delle cellule di Leydig e di altri ormoni, tra cui l’inibina, da parte delle cellule di Sertoli.
Il principale androgeno prodotto dai testicoli è il testosterone, ma vi sono prodotti anche l’androstenedione e il deidro-epiandrosterone solfato (DHEAS). Il diidro-testosterone (DHT) è il metabolita attivo del testosterone, derivato dalla conversione periferica del testosterone mediata dall’enzima 5a-reduttasi.
Testosterone e DHT svolgono un’importante azione virilizzante, mediante l’attivazione del recettore degli androgeni (AR) e sono responsabili dello sviluppo dei caratteri sessuali secondari.
Le cellule di Sertoli del testicolo producono l’inibina B, che aumenta durante la minipuberty e poi durante la pubertà. Nell’età adulta l’inibina B esercita un feed-back negativo sull’FSH e riflette la spermatogenesi. L’ormone anti-mulleriano (AMH) è secreto dalle cellule di Sertoli durante la vita fetale e causa la regressione dei dotti di Muller (fig. 1).
Attualmente non sono ancora completamente noti i meccanismi che inducono l’attivazione a livello dell’asse HPG, anche se viene ipotizzato che una complessa interazione di neurotrasmettitori controlli il SNC e che le variazioni di fattori inibenti e stimolanti inducano l’attivazione dell’asse HPG al momento della pubertà. Tra i meccanismi associati all’attivazione del GnRH, uno di recente descrizione è quello regolato dalla leptina. La leptina è un ormone prodotto dagli adipociti in concentrazioni proporzionali ai depositi adiposi; il suo fisiologico aumento in corrispondenza della pubertà potrebbe segnalare il raggiungimento di depositi energetici critici per l’innescarsi di questi processi evolutivi. Un altro fattore che gioca un ruolo centrale per la secrezione di GnRH è la kiss-peptina, un neuropeptide prodotto dall’ipotalamo. Mutazioni che causano una perdita di funzione del gene del recettore della kiss-peptina (Kiss1R) causano ipogonadismo ipogonadotropo.
Solitamente circa due anni prima dell’attivazione gonadica, quindi intorno ai 7-8 anni nel maschio, si osservano modificazioni nella secrezione surrenalica di DHEA, DHEAS e androstenedione in un fenomeno noto come adrenarca, che contribuisce in maniera indipendente allo sviluppo dei caratteri sessuali secondari rispetto all’attivazione gonadica. Talvolta questa attivazione surrenalica avviene un poco prima e spesso comporta la comparsa precoce di peluria pubica e/o ascellare (adrenarca e/o pubarca precoce).

Figura 1 (modificata da 3)
Rappresentazione schematica dei livelli circolanti nel maschio di gonadotropine, testosterone (prodotto dalle cellule di Leydig), inibina e AMH (prodotti dalle cellule di Sertoli)
Tappe dello sviluppo puberale nel maschio
Durante la pubertà avviene una serie di cambiamenti fisici che comprendono aumento della velocità di crescita, modifica della composizione corporea a favore della massa muscolare, sviluppo dei caratteri sessuali primari e secondari.
L’aumento delle dimensioni testicolari è il primo segno di reale attivazione puberale nel maschio. Spesso, però, i segni più tangibili dello sviluppo puberale in corso sono i caratteri sessuali secondari: aumento delle dimensioni del pene, comparsa di peluria pubica e ascellare, baffi, barba e cambiamento del tono di voce.

Figura 2 (modificata da 3)
Evoluzione dello sviluppo testicolare dalla nascita all’età adulta. I tubuli seminiferi (cellule di Sertoli + cellule germinali) sono il maggiore componente dei testicoli. Durante tutto il periodo pre-puberale (stadio I di Tanner) il volume dei tubuli seminiferi dipende principalmente dalle cellule di Sertoli, mentre l’aumento delle dimensioni testicolari durante lo sviluppo puberale (stadio II-V di Tanner) è principalmente dovuto alla proliferazione delle cellule germinali
L’aumento della velocità di crescita (spurt puberale) dipende principalmente dall’interazione degli steroidi gonadici con il GH: nel maschio varia da 6 cm/anno a 10-15 cm/anno e avviene principalmente nelle fasi intermedie della pubertà (stadi puberali G3-G4, vedi oltre).
Nel periodo puberale è comune la ginecomastia, dovuta alla proliferazione del tessuto ghiandolare e fibroso della mammella. Può essere monolaterale o bilaterale ed è solitamente transitoria, con tendenza alla regressione spontanea nell’arco di alcuni mesi/anni. La ginecomastia è dovuta allo sbilanciamento tra estrogeni e androgeni a favore dei primi, a causa dell’importante attività steroidogenica del periodo puberale e/o per l’aumentata attività locale dell’aromatasi (enzima che converte gli androgeni in estrogeni). In alcuni casi, tuttavia, la ginecomastia è espressione di una patologia sottostante, come la sindrome di Klinefelter o l’iperprolattinemia. Nel caso in cui non vi sia il sospetto di una patologia sottostante, bisogna rassicurare il ragazzo e i genitori sulla transitorietà della ginecomastia e, solo nei casi di ginecomastia persistente e inficiante le attività sociali del ragazzo, può essere indicata un approccio terapeutico.
Valutazione dello sviluppo puberale nel maschio
Per valutare in maniera oggettivabile la progressione dello sviluppo puberale è stata creata una classificazione, detta stadiazione di Tanner (fig. 3), che considera lo sviluppo dei genitali (G1-5) e della peluria pubica (P1-5). Solitamente l’aumento delle dimensioni testicolari precede la comparsa di peluria pubica, ma esiste una grande variabilità inter-individuale. La classificazione attribuisce lo stadio 1 ai soggetti prepuberi e gli stadi 4-5 ai soggetti adulti.
La stadiazione di Tanner si basa sulla stima del volume testicolare mediante la palpazione dei testicoli e il confronto visivo con l’orchimetro di Prader (uno strumento costituito da una serie di modelli di forma ellissoide aventi un volume crescente da 1 a 25 mL) e sulla valutazione della peluria pubica. L’aumento delle dimensioni testicolari da 2-3 mL tipiche dell’età infantile a > 3 mL indica il passaggio dallo stadio pre-pubere al primo stadio dell’inizio della pubertà (G2).
Stadi per i genitali (G):
- stadio G1 (pre-adolescente): testicoli, scroto e pene presentano aspetto infantile;
- stadio G2: scroto e testicoli aumentano di dimensioni (> 3 mL) e la cute dello scroto inizia a diventare più scura;
- stadio G3: si assiste alla crescita del pene, sia in lunghezza che in larghezza, e a un’ulteriore crescita dei testicoli (5-10 mL) e dello scroto;
- stadio G4: il pene aumenta ulteriormente in lunghezza e larghezza, con maggiore sviluppo del glande; i testicoli (10-15 mL) e lo scroto aumentano; la cute dello scroto diventa più iperpigmentata;
- stadio G5: i genitali raggiungono le dimensioni da adulto.
Stadi per la peluria pubica (P):
- stadio P1 (pre-adolescente): assenza di peli pubici;
- stadio P2: peluria pubica leggermente pigmentata, lanuginosa, liscia o solo leggermente arricciata, localizzata soprattutto alla base del pene;
- stadio P3: peli pubici più scuri, spessi e arricciati, con maggiore diffusione in sede pubica;
- stadio P4: peluria pubica di tipo adulto, ma in un’area più piccola rispetto a quella dello stadio da adulto (senza diffusione alla superficie mediale delle cosce);
- stadio P5: peli pubici di tipo adulto come quantità e tipologia, distribuiti su tutta l’area pubica e diffusi alla superficie mediale delle cosce.

Per completare la valutazione puberale, occorre anche considerare la comparsa di peluria ascellare e al volto, l’abbassamento del tono di voce e la prima eiaculazione; questi ultimi due avvengono solitamente nella fase intermedia dello sviluppo, a un’età compresa tra 13 e i 14 ½ anni.
L’aumento delle dimensioni testicolari è dovuto alla crescita dei tubuli seminiferi ed è un marcatore indiretto della spermatogenesi.
Per valutare lo sviluppo fisiologico non è in genere necessaria l’ecografia testicolare, che tuttavia può essere un utile strumento diagnostico per valutare in modo più accurato le dimensioni dei testicoli, distinguendo tra tessuto testicolare e tessuto circostante. La misurazione di lunghezza, ampiezza e profondità permette di calcolare il volume testicolare (V = l x a x p x π/6). Inoltre, mediante l’esame ecografico è possibile rilevare alcune condizioni patologiche, tra cui tumori testicolari (in particolare i tumori a cellule germinali e i tumori a cellule di Leydig), microlitiasi testicolare, idrocele e varicocele.
Il dosaggio delle gonadotropine basali e degli androgeni fornisce una valutazione dell’asse HPG. All’inizio dello sviluppo puberale i valori di gonadotropine basali sono scarsamente informativi, per cui nel caso si sospetti un anticipo o un ritardo puberale, può essere utile effettuare un test da stimolo con GnRH o analogo. Oltre alla misurazione delle gonadotropine, è utile valutare gli androgeni, in particolare il testosterone.
Bibliografia
- Tinggaard J, et al. The physiology and timing of male puberty. Curr Opin Endocrinol Diabetes Obes 2012, 19: 197-203.
- Marshall WA, Tanner JM. Variations in the pattern of pubertal changes inboys. Arch Dis Child 1970, 45: 13-23.
- Rey RA, et al. Are Klinefelter boys hypogonadal? Acta Paediatr 2011, 100: 830-8.
Overview sugli ipostaturismi
Chiara Maggioli
Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
Definizione e cause
Si definisce bassa statura un’altezza inferiore a 2 deviazioni standard (< 3° percentile) rispetto all’altezza media di individui di pari età e sesso (e possibilmente etnia)(1). La bassa statura può essere una variante fisiologica di crescita oppure essere espressione di una patologia sottostante.
| Cause di bassa statura (1,2) | |||
| Fisiologiche | Bassa statura familiare Ritardo costituzionale di crescita e adolescenza Forme miste |
||
| Patologiche | Genetiche | Sindromi | Turner Down Noonan Silver-Russel Prader-Willi Neurofibromatosi Di George |
| Mutazione del gene SHOX | |||
| Endocrine | Difetti dell’asse GH-IGF-I Ipotiroidismo Ipercortisolismo Ipoparatiroidismo Pseudoipoparatiroidismo Disordini del metabolismo o dell’azione della vitamina D Diabete mellito scarsamente controllato |
||
| Patologie scheletriche | Osteocondrodisplasie | ||
| SGA senza catch-up growth | |||
| Malattie e infezioni croniche | Cardiache Renali Onco-ematologiche Metaboliche Polmonari Infezioni |
||
| Malnutrizione | Diminuito introito Malassorbimento |
||
| Fattori psico-sociali | |||
| Iatrogene | Farmaci Irradiazione |
||
La bassa statura di cui non si riconosce alcuna causa viene definita bassa statura idiopatica.
Nella pratica clinica la valutazione di un paziente con bassa statura deve prevedere un’accurata anamnesi personale e familiare e un attento esame obiettivo, volti a riconoscere precocemente e trattare la cause patologiche di tale condizione.
Approccio al bambino con bassa statura (3,4)
I dati anamnestici (2) più significativi sono:
- altezza ed età di sviluppo dei genitori ed eventualmente di fratelli o sorelle;
- parametri neonatali di peso, lunghezza e circonferenza cranica;
- decorso del periodo neonatale;
- anamnesi patologica e farmacologica del bambino ed eventuale identificazione di condizioni psico-sociali che possano alterarne la crescita;
- raccolta di dati sulla crescita staturale e ponderale precedente all’osservazione clinica.
L’altezza dei genitori permette di definire il target familiare mediante la formula (tutti i dati in cm):
- femmine: (altezza della madre + altezza del padre - 13)/2
- maschi: (altezza della madre + altezza del padre + 13)/2
Se possibile, è opportuno misurare direttamente la mamma e il papà, poiché le altezze riferite mediamente sono sovrastimate. Il target familiare ci permette di identificare il centile lungo il quale dovrebbe crescere il bambino. Questo dato è determinante per la diagnosi di bassa statura familiare e fornisce un’indicazione importante laddove invece l’altezza del bambino, seppure nel range di normalità, si discosti significativamente dal centile genitoriale.
I parametri neonatali vanno rapportati all’età gestazionale e sono indicatori della crescita in utero, che dipende principalmente dal corretto funzionamento dell’unità feto-placentare. Peso e/o lunghezza < 3° centile alla nascita permettono di identificare il bambino SGA (small for gestational age).
Il decorso del periodo neonatale può fornire informazioni importanti nella valutazione della bassa statura: ad esempio, la presenza di ipoglicemia e ittero prolungato può associarsi al deficit di GH.
Tra le cause di bassa statura bisogna inoltre considerare in anamnesi le patologie croniche e aspetti psico-sociali che possano influenzare la crescita.
Se possibile, è opportuno cercare di ottenere il maggior numero possibile di dati riguardanti la crescita del bambino: è infatti di fondamentale importanza capire se si tratta di una crescita lenta ma costante nel tempo o di un vero e proprio arresto nella velocità di crescita.
L’esame obiettivo (2) del bambino con bassa statura deve prevedere:
- misurazione accurata della lunghezza/altezza e peso (e circonferenza cranica nei bambini < 2 anni);
- valutazione delle proporzioni corporee e dei parametri antropometrici;
- valutazione dello sviluppo puberale;
- ricerca di segni dismorfici che possano indirizzare verso l’identificazione di quadri sindromici;
- ricerca di segni/sintomi compatibili con malattie sistemiche o con cause endocrinologiche di bassa statura;
- valutazione della velocità di crescita in un periodo di almeno 6 mesi.
La parte principale dell’esame obiettivo consiste in un’accurata valutazione dei parametri di crescita. Fino all’età di due anni il bambino viene misurato disteso per definire la lunghezza; dopo i due anni la misurazione avviene in piedi. L’altezza è la media di 3 misurazioni effettuate estendendo la colonna con una lieve trazione in alto della mastoide al termine dell’espirazione. L’altezza e il peso sono quindi riportati su una curva di crescita e si identifica il percentile o la deviazione standard a cui corrispondono e la proporzione dell’altezza con il peso.
Oltre ai parametri misurati in uno specifico momento, è di fondamentale importanza il calcolo della velocità di crescita nel tempo, che generalmente viene calcolato in cm/anno sulla base di due misurazioni che distano almeno 6 mesi. La velocità di crescita va valutata in base all’età del paziente, mediante curve di percentili che tengono conto dell’età e dello sviluppo puberale. Una velocità di crescita normale, anche in un bambino di bassa statura, permette di escludere la maggior parte delle cause patologiche di difetti di crescita.
Nell’esame obiettivo è opportuno misurare la circonferenza cranica nei bambini di età < 2 anni, l’altezza in posizione seduta, l’apertura delle braccia e la lunghezza degli arti inferiori per valutare le proporzioni corporee.
La valutazione dello sviluppo puberale si avvale delle tabelle di Tanner e permette di identificare i periodi di maggiore o minore velocità di crescita staturale non solo in base all’età del bambino, ma con più precisione, in base all’avanzamento della pubertà. Nella fase prepuberale è, infatti, normale osservare un rallentamento della velocità di crescita, che invece aumenta raggiungendo un picco all’inizio della pubertà nelle femmine e allo stadio 2-3 di Tanner nei maschi.
L’esame obiettivo generale permette di identificare tratti somatici suggestivi di condizioni sindromiche associate a bassa statura o caratteristiche cliniche indicative di malattie sistemiche di origine endocrina o non endocrina.
Esami diagnostici di primo livello
Accertata la presenza di bassa statura o di un effettivo rallentamento della velocità di crescita, è opportuno eseguire:
- radiografia mano-polso sinistro per età ossea
- indagini di laboratorio.
La radiografia mano-polso consente di identificare l’età ossea del bambino; generalmente il metodo più usato nell’interpretazione è il confronto con l’atlante di Greulich and Pyle. L’età ossea è un elemento importante per la diagnosi differenziale delle cause di bassa statura e permette di eseguire il calcolo della previsione staturale.
Tra gli esami di laboratorio di primo livello è opportuno eseguire: emocromo, funzione epatica e renale, VES, esame urine, screening per la celiachia, funzione tiroidea, IGF-I. Eventuali ulteriori approfondimenti specifici dovrebbero essere eseguiti sulla base di uno specifico sospetto clinico. In particolare nelle bambine, escluse le cause più frequenti di bassa statura, è opportuno eseguire l’esame del cariotipo per escludere la sindrome di Turner.

Bibliografia
- Dattani M, Preece M. Growth hormone deficiency and related disorders: insights into causation, diagnosis, and treatment. Lancet 2004, 363: 1977-87.
- Rogol AD, Hayden GF. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr 2014, 164 (5 Suppl): S1-14.
- Cohen P, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab 2008, 93: 4210–7.
- Rogol AD, Geffner M, Hoppin AG. Diagnostic approach to short stature. UpToDate.
Ipostaturismi dipendenti da alterazioni dell'asse GHRH-GH-IGF
Chiara Maggioli
Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
EPIDEMIOLOGIA E CLASSIFICAZIONI
La bassa statura associata a deficit di GH (GHD) ha un’incidenza di circa 1:4000-1:10.000. Spesso la diagnosi di GHD risulta molto complessa, poiché ad oggi non esistono esami di laboratorio o test di stimolo che rappresentino il gold standard diagnostico. Probabilmente inoltre, considerando che il 25-75% dei pazienti risulta normale a un successivo test al termine della crescita, spesso il GHD risulta sovra-diagnosticato (1). Il criterio più valido, in ogni caso, più delle indagini di laboratorio, è l’osservazione clinica del paziente e dei parametri auxologici (2). Circa il 3-30% dei GHD è una forma familiare. Tra le cause di GHD si distinguono cause genetiche, organiche oppure idiopatiche. Dal punto di vista clinico, la classificazione distingue le forme in congenite e acquisite.
| Classificazione eziologica del GHD (1) | |||
| Congenito | Genetico | Deficit isolato asse GHRH-GH-IGF | Mutazioni del recettore del GHRH Mutazioni o delezioni del gene GH1 Mutazioni che causano insensibilità all’azione del GH |
| Difetti ipofisari multipli | Mutazioni di POU1F1 Mutazioni di PROP1 Mutazioni di LHX3 Mutazioni di LHX4 Mutazioni di TBX19 (TPIT) Mutazioni di SOX3 Mutazioni di SOX2 Mutazioni di HESX1 |
||
| Associato a difetti strutturali cerebrali | Agenesia del corpo calloso Displasia setto-ottica Oloprosencefalia Encefalocele Idrocefalo |
||
| Associato ad anomalie della linea mediana | Labio-palatoschisi Incisivo centrale unico |
||
| Acquisito | Traumi | Perinatali Post-natali |
|
| Infezioni e infiammazioni | Meningiti Encefaliti |
||
| Tumori | Craniofaringiomi Germinomi Adenomi ipofisari Gliomi ottici |
||
| Istiocitosi a cellule di Langerhans | |||
| Iatrogeno | Irradiazione cranica Chemioterapia |
||
| Emorragie intra-craniche | |||
| Ipotiroidismo | |||
| Deprivazione psico-sociale | |||
| Disfunzione neuro-secretoria | |||
| Idiopatico | |||
Tra le cause genetiche sono state descritte in letteratura mutazioni autosomico dominanti, autosomico recessive e legate al cromosoma X.
Si definisce come deficit di GH idiopatico una condizione caratterizzata da un deficit di GH confermato dai test di stimolo in assenza di causa nota. Un recente studio condotto su 38 bambini ha in realtà posto in evidenza come più del 90% dei pazienti identificati come affetti da GHD idiopatico (risposta patologica sub-normale a due test di stimolo e RMN negativa) siano risultati normali a un retesting precoce. Questo permette di ipotizzare come la diagnosi di GHD idiopatico spesso sia dovuta a falsi positivi dei test di stimolo, soprattutto nei bambini dove la risposta in termini di recupero di crescita non sia ottimale dopo l’inizio della terapia con GH (3).
PRESENTAZIONE CLINICA
La presentazione clinica del GHD dipende dall’età di insorgenza del difetto e dalla presenza di altri deficit ipofisari associati.
GHD congenito
L’esordio neonatale ha come principali caratteristiche l’ipoglicemia, l’ittero prolungato, il micropene. Il neonato non presenta deficit staturale importante e il rallentamento nella velocità di crescita avviene progressivamente dopo i primi 2 anni di vita (1). L’ipoglicemia tipicamente è più severa se al deficit di GH si associa deficit di ACTH. Spesso in anamnesi è riportato un trauma da parto.
GHD acquisito
Il bambino con GHD presenta un severo rallentamento della velocità di crescita, bassa statura armonica, età ossea ritardata con ritardo nella chiusura delle fontanelle, distribuzione del tessuto adiposo prevalentemente facio-cervico-tronculare, facies “angelica” o “da bambola” con tratti somatici poco marcati, naso a sella con iposviluppo del ponte nasale e bozze frontali prominenti. La voce è tipicamente infantile, i capelli spesso radi e sottili, vi può essere microgenitosomia con ritardo nello sviluppo puberale.
Le caratteristiche cliniche che devono indirizzare la diagnosi di una bambino con bassa statura verso il sospetto di deficit di GH sono (4):
- bassa statura: < -2.5 DS;
- ridotta velocità di crescita: < -1.5 DS o < 25° centile;
- ritardo dell’età ossea: > 1 anno.
Criteri che necessitano una valutazione immediata per GHD sono:
- bassa statura: < -3 DS;
- statura < -1.5 DS sotto quella prevista in base al target familiare;
- statura < -2 DS con velocità di crescita < -1 DS;
- velocità di crescita < -2 DS calcolata in un anno o < -1.5 DS se per più di due anni;
- segni o sintomi di lesione intra-cranica;
- segni o sintomi di deficit tropinici multipli;
- segni o sintomi neonatali di GHD.
DIAGNOSI
IGF-I e IGF-BP3
Dopo avere escluso le cause più frequenti di bassa statura o nel caso di sospetto clinico di GHD, è opportuno eseguire come esami di primo livello il dosaggio di IGF-I e IGF-BP3. Al contrario del GH, queste sono stabili nel tempo, pertanto è preferibile questo dosaggio rispetto a quello del GH basale. Valori bassi di IGF-I e IGF-BP3 possono indirizzare nella diagnosi di GHD, ma da soli hanno bassa specificità e sensibilità (1).
Le IGF-I, infatti, sono relativamente basse nell’infanzia, con un range di normalità che si sovrappone parzialmente al range considerato sospetto per GHD. Inoltre alimentazione inadeguata, problemi epatici, renali e la presenza di patologia neoplastica possono ridurre i valori di IGF-I in assenza di GHD (2).
Le IGF-BP3 sono le principali proteine leganti le IGF-I, sono meno dipendenti dall’apporto nutrizionale e possono essere più discriminatorie rispetto alle IGF-I per valori bassi della norma, soprattutto nei bambini più piccoli. Nei pazienti con GHD severo, comunque, IGF-I e IGF-BP3 sono quasi sempre ridotte.
Il valore di IGF-I deve essere interpretato in base a età, sesso e stadio puberale: si considerano suggestivi per GHD livelli di IGF-I < -2 DS.
Test di stimolo (2)
Per la diagnosi di GHD sono comunemente utilizzati diversi test che valutano la risposta del GH allo stimolo: test con arginina, clonidina, glucagone, insulina o il test combinato con GHRH + arginina. I test di stimolo devono essere eseguiti la mattina a digiuno. L’infusione del farmaco scelto sarà seguita da prelievi per GH a intervalli stabiliti. Prima del test occorre accertarsi, nel sospetto di deficit ipofisari multipli, che il resto della funzione ipofisaria sia ben compensata.
Limiti dei test di stimolo farmacologici:
- i test non sono fisiologici;
- il cut-off di normalità è arbitrario;
- la risposta del GH al test dipende dall’età del paziente, dallo stadio puberale (risposta ridotta in fase pre-puberale) e dall’indice di massa corporea (risposta ridotta nei bambini obesi);
- i metodi di dosaggio del GH presentano diversa accuratezza diagnostica;
- i test sono scarsamente riproducibili: come dimostra lo studio di Loche (5), la maggior parte dei bambini definiti GHD con 2 test patologici, in assenza di anomalie strutturali, presenterà una normale risposta a un terzo test eseguito a distanza di 1-6 mesi.
Secondo le raccomandazioni dell’Istituto Nazionale per l’Eccellenza Clinica (NICE) britannico è opportuno eseguire almeno due test e porre diagnosi di GHD laddove siano entrambi patologici. Attualmente in Italia si considera patologica una risposta (nota 39) se il GH risulta < 8 µg/L in due test di stimolo classici eseguiti in due giorni differenti (se il test usato è GHRH + arginina, il cut-off è < 20 µg/L). Sebbene molto raramente, una normale risposta del GH ai test classici, in presenza di un reale rallentamento di crescita con bassi livelli di IGF-I e IGF-BP3, può essere indicativa di una condizione definita come disfunzione neuro-secretoria(1).
In conclusione, la variabile risposta ai test, la loro non riproducibilità e l’arbitrarietà dei cut-off rendono piuttosto complessa la diagnosi di GHD, soprattutto in quei pazienti in cui le indagini radiologiche non mostrano anomalie strutturali ipotalamo-ipofisarie (1).
Peri-pubertà
Durante il periodo peri-puberale, fisiologicamente la risposta ai test di stimolo è ridotta, per cui alcuni autori consigliano di eseguire un priming prima del test mediante la somministrazione di testosterone nel maschio ed estrogeni nella femmina. In realtà non c'è un consenso in letteratura sulla necessità di eseguire il priming in tutti i pazienti nè sul protocollo da utilizzare (4).
Prima infanzia
Al di sotto dei 2 anni di vita non è necessario praticare i test farmacologici se la RM ha dimostrato un’anomalia dell’adeno-ipofisi associata a quella del peduncolo e/o della neuro-ipofisi in un bambino con decelerazione della velocità di crescita o segni clinici riferibili a ipopituitarismo e/o ipoglicemia (nota 39).
Diagnostica radiologica
In tutti i pazienti in cui è posta diagnosi clinica e biochimica di GHD, è essenziale eseguire una risonanza magnetica cerebrale e nello specifico della regione ipotalamo-ipofisaria con mezzo di contrasto. La RMN permette di valutare la presenza di lesioni espansive o di anomalie morfologiche, quali ipoplasia dell’adeno-ipofisi, agenesia del peduncolo, ectopia della neuro-ipofisi. L’imaging patologico è un elemento importante per la diagnosi, soprattutto nell’identificare i GHD permanenti (6).
TERAPIA DEL DEFICIT DI GH
Non appena viene posta diagnosi di GHD, dopo l’esecuzione delle indagini strumentali, è opportuno intraprendere la terapia con ormone della crescita ricombinante. La precocità del trattamento è, infatti, correlata con una migliore risposta in termini di accrescimento. Mediamente il guadagno staturale è di circa 30 cm nei GHD severi, ma tale valore è ampiamente influenzato dalla dose, dall’età di insorgenza del deficit e dalla precocità di inizio della terapia (1).
Obiettivi della terapia:
- aumentare la velocità di crescita staturale;
- aumentare la statura adulta;
- modificare la composizione corporea;
- determinare effetti metabolici positivi;
- migliorare la qualità della vita.
Modalità di somministrazione e dosi
Il GH ricombinante è somministrato mediante iniezioni sottocutanee giornaliere per 6-7 giorni a settimana (la somministrazione quotidiana è necessaria quando il GHD si associa a difetti pluritropinici per il rischio maggiore di ipoglicemie).
La dose media è di 25-50 µg/kg/die, ma solitamente le dosi utilizzate sono nel range più basso e comunque è consigliabile utilizzare la dose minima efficace.
Monitoraggio e follow-up
Il monitoraggio è principalmente clinico: soprattutto nei primi due anni l’efficacia del trattamento, che riflette anche la correttezza della diagnosi, è dimostrata da un importante incremento della velocità di crescita. Inoltre, viene eseguito di routine il dosaggio di IGF-I e IGF-BP3, soprattutto per valutare la compliance e la sicurezza della terapia. Nei bambini con anomalie ipofisarie complesse occorre inoltre valutare, dopo l’introduzione della terapia con GH, la funzione ipofisaria, poiché tale terapia può slatentizzare altri deficit tropinici prima non diagnosticati o può rendere necessario l’incremento della dose sostitutiva.
Al raggiungimento della statura definitiva (velocità di crescita < 2 cm/anno), la terapia con GH viene sospesa per eseguire un retesting dopo 1-3 mesi. Nel caso di deficit genetico accertato o panipopituitarismo, la terapia viene proseguita senza interruzioni. Il retesting viene eseguito mediante test con insulina o GHRH + arginina: è considerato patologico un picco di GH < 6 µ/L dopo ipoglicemia insulinica o < 19 µ/L dopo test con GHRH + arginina. In caso di positività del test, si ricomincia la terapia con GH a un dosaggio appropriato per l’età. Per i criteri dettagliati di prescrivibilità si rimanda alla nota 39.

Effetti collaterali
- Arrossamento e prurito in sede di iniezione.
- Ipertensione endocranica benigna (più frequente nelle prime settimane di terapia, ma può insorgere anche tardivamente; è risolta dalla sospensione della terapia che può essere reintrodotta gradualmente senza rischio aumentato di recidiva).
- Ginecomastia prepuberale.
- Artralgia, edema, mialgia (più frequentemente riscontrati negli adulti).
- Insulino-resistenza.
- Epifisiolisi della testa del femore (l’aumentata frequenza di epifisiolisi si verifica solo in bambini con GHD da causa organica, non in quelli con GHD idiopatico)(1).
È ancora molto dibattuto se la terapia a lungo termine con GH aumenti il rischio di neoplasie o di recidive in caso di precedenti tumori. Alcuni dati sembrano indicare un’incidenza aumentata di tumori, ma le dosi di GH in quei pazienti erano al di sopra dei range giornalieri normalmente utilizzati. A tale riguardo è attualmente in corso in Europa lo studio SAGhE (Safety and Appropriateness of Growth hormone treatments in Europe), i cui dati definitivi non sono ancora disponibili. Per quanto riguarda le recidive di neoplasia, la terapia con GH non sembrerebbe aumentare il rischio (1), ma si consiglia di non iniziare il trattamento nel caso di malattia attiva e nei primi 1-2 anni di follow-up.
Un recente studio ha evidenziato l’aumentato rischio di ictus emorragico in età adulta in pazienti trattati con GH nell’infanzia (7). In realtà alcune società scientifiche internazionali hanno messo in evidenza i forti limiti di quello studio, per cui attualmente non c’è stata alcuna indicazione alla revisione riguardo la prescrivibilità, né la necessità di eseguire follow-up specifico per tale complicanza.
BIBLIOGRAFIA
- Dattani M, Preece M. Growth hormone deficiency and related disorders: insights into causation, diagnosis, and treatment. Lancet 2004, 363: 1977-87.
- Stanley T. Diagnosis of growth hormone deficiency in childhood. Curr Opin Endocrinol Diabetes Obes 2012, 19: 47–52.
- Bizzarri C, Pedicelli S, Boscherini B, et al. Early retesting by GHRH + arginine test shows normal GH response in most children with idiopathic GH deficiency.J Endocrinol Invest 2014, DOI: 10.1007/s40618-014-0205-3.
- GH Research Society consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. J Clin Endocrinol Metab 2000, 85: 3990-3.
- Loche S, Bizzarri C, Maghnie M, et al. Results of early reevaluation of growth hormone secretion in short children with apparent growth hormone deficiency. J Pediatr 2002, 140: 445–9.
- Coutant R, Rouleau S, Despert F, et al. Growth and adult height in GH-treated children with nonacquired GH deficiency and idiopathic short stature: the influence of pituitary magnetic resonance imaging findings. J Clin Endocrinol Metab 2001, 86: 4649–54.
- Poidvin A, Touzé E, Ecosse E, et al. Growth hormone treatment for childhood short stature and risk of stroke in early adulthood. Neurology 2014, 83: 780-6.
Ipostaturismi endocrini non GH-dipendenti
Chiara Maggioli
Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
I disturbi endocrini che possono determinare bassa statura sono piuttosto rari, ma è fondamentale il loro riconoscimento, perché possono sottendere patologie severe e rientrano per lo più tra le cause trattabili di bassa statura (1). In generale ma non sempre, il deficit di crescita da causa endocrinologica si associa a incremento ponderale.
Ipotiroidismo
Rappresenta una delle cause più frequenti di ipostaturismo endocrino. Può insorgere in qualunque fase della crescita del bambino e le sue manifestazioni cliniche sono variabili in funzione dell’età di esordio. L’ipotiroidismo può essere centrale o primitivo, congenito o acquisito. Nel caso di ipotiroidismo centrale, è opportuno valutare la presenza di eventuali altri deficit tropinici.
Dal punto di vista clinico, si può osservare un rallentamento della velocità di crescita con età ossea ritardata, associato a segni e sintomi caratteristici dell’ipotiroidismo legati alla severità del quadro. È opportuno in ogni bambino valutato per bassa statura eseguire dosaggio di TSH ed fT4.
Ipertiroidismo
Raramente bambini con ipertiroidismo cronico non trattato possono presentare un pattern di crescita caratterizzato da incremento della velocità di crescita staturale, che si accompagna a precoce maturazione delle epifisi, con conseguente bassa statura finale (2).
Sindrome di Cushing
L’eccesso di glucocorticoidi può essere talvolta associato ad eccesso di androgeni surrenalici. Si distinguono forme endogene e forme caratterizzate da eccesso di glucocorticoidi esogeni. Le principali cause di Cushing endogeno nei bambini sono rappresentate da adenomi ipofisari ACTH-secernenti, adenomi e carcinomi cortico-surrenalici, variabilmente distribuiti per incidenza in base all’età dei pazienti.
Dal punto di vista clinico, il ritardo o arresto di crescita si associa a incremento ponderale diffuso: questi possono essere gli unici segni presenti alla diagnosi. Possono essere variabilmente associati altri segni e sintomi caratteristici della sindrome di Cushing: obesità centrale, gibbo, strie rubre, irsutismo, ipotrofia muscolare, acne e disturbi neuropsicologici (3).
I test migliori per la diagnosi sono il dosaggio del cortisolo libero urinario e il test di soppressione con desametasone overnight.
La terapia permette di ottenere un ottimo catch-up growth nella maggior parte dei pazienti.
Pubertà precoce
È una condizione caratterizzata dall’inizio dello sviluppo puberale in età < 8 anni nelle femmine e < 9 anni nei maschi. Gli steroidi sessuali determinano uno sviluppo epifisario accelerato, con conseguente aumento della velocità di crescita ed avanzamento ancora più rapido dell’età ossea. Di conseguenza, sebbene nella fase di sviluppo questi bambini presentino una crescita maggiore rispetto alla media per età, la chiusura delle epifisi avviene in epoca precoce, determinando una bassa statura in età adulta(4).
La terapia di tali condizioni dipende dalla causa sottostante, ma in ogni caso ha come obiettivo quello di bloccare il processo puberale per posticiparlo ad un’età fisiologica.
Diabete mellito di tipo 1
Il diabete mal controllato dalla terapia medica può determinare rallentamento della crescita staturale per deficit calorico dovuto principalmente alla glicosuria (5). I bambini con DM1 possono avere alterazione nella sintesi o azione delle IGF-I e alcuni autori descrivono una correlazione inversa tra emoglobina glicata e statura finale (6). Attualmente è raro che si verifichino tali circostanze e i bambini in terapia per DM1 raggiungono per lo più una statura da adulti appropriata al target familiare.
Patologie endocrine associate ad alterazioni scheletriche
Alterazioni della vitamina D. Tutti i disordini dovuti ad alterazioni nella sintesi, metabolismo o azione della vitamina D possono determinare rachitismo, caratterizzato da diversi segni clinici tra cui alterato sviluppo delle epifisi, incurvamento delle ossa lunghe e conseguente rallentamento della crescita e bassa statura.
Pseudoipoparatiroidismo e osteodistrofia di Albright. È una condizione patologica caratterizzata da resistenza all’azione del PTH, con conseguente ipocalcemia e iperfosforemia in presenza di valori elevati di PTH. Esistono diverse forme di pseudoipoparatiroidismo, che variano per modalità di trasmissione genetica e di espressione clinica (7). Alcune forme di pseudoipoparatiroidismo si associano al quadro definito osteodistrofia di Albright, che si caratterizza per:
- obesità addominale
- bassa statura
- facies rotondeggiante “a luna piena”
- brevità del 4° metacarpo
- ritardo mentale variabile.
Bibliografia
- Rogol AD, Hayden GF. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr 2014, 164 (5 Suppl): S1-14.
- Conwell LS. Graves' disease--acceleration of linear growth. J Pediatr Endocrinol Metab 2009, 22: 289-90.
- Magiakou MA, Mastorakos G, Oldfield EH, et al. Cushing's syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med 1994, 331: 629-36.
- Biro FM, McMahon RP, Striegel-Moore R, et al. Impact of timing of pubertal maturation on growth in black and white female adolescents: The National Heart, Lung, and Blood Institute Growth and Health Study. J Pediatr 2001, 138: 636-43.
- Mauras N, Merimee T, Rogol AD. Function of the growth hormone-insulin-like growth factor I axis in the profoundly growth-retarded diabetic child: evidence for defective target organ responsiveness in the Mauriac syndrome. Metabolism 1991, 40: 1106-11.
- Bonfig W, Kapellen T, Dost A, et al. Growth in children and adolescents with type 1 diabetes. J Pediatr 2012, 160: 900-3.
- Levine MA. An update on the clinical and molecular characteristics of pseudohypoparathyroidism. Curr Opin Endocrinol Diabetes Obes 2012, 19: 443-51.
Ipostaturismi non endocrini
Chiara Maggioli
Dipartimento di Fisiopatologia Clinica, Unità di Endocrinologia, Università di Firenze
Gli ipostaturismi da causa non endocrina si distinguono in condizioni fisiologiche e patologiche.
CAUSE FISIOLOGICHE
Rappresentano le cause più frequenti di bassa statura (1).
Bassa statura familiare: si caratterizza per anamnesi familiare positiva per bassa statura, velocità di crescita nella norma, età ossea coincidente con l’età cronologica e sviluppo puberale in epoca fisiologica. È una condizione che non richiede alcun trattamento e che determinerà una statura definitiva appropriata al target familiare.
Ritardo costituzionale di crescita e adolescenza: si caratterizza per normale lunghezza alla nascita, con riduzione della velocità di crescita nei primi 3-5 anni. Il bambino successivamente cresce lungo i centili inferiori della norma per altezza, con una velocità di crescita sostanzialmente normale. L’età ossea è ritardata rispetto all’età cronologica. In fase pre-puberale si osserva una riduzione della velocità di crescita e tipicamente si assiste a un ritardo nello sviluppo puberale. Spesso l’anamnesi familiare è positiva per ritardo di sviluppo. La statura finale sarà adeguata al target familiare.
Forme miste: le forme in cui si ha associazione tra bassa statura familiare e ritardo costituzionale di crescita sono di difficile diagnosi differenziale con il GHD, potendo mostrare importante difetto staturale associato ad età ossea ritardata. Anche in questo caso la statura definitiva sarà appropriata al target familiare.
CAUSE PATOLOGICHE
Sindromi genetiche (Turner, Down, Noonan, Silver-Russel, Prader-Willi, neurofibromatosi, Di George). Il riconoscimento precoce delle sindromi associate a bassa statura permette un corretto inquadramento del paziente e la possibilità di monitorare e trattare eventuali comorbilità.
Tra le varie sindromi genetiche associate a bassa statura, in Italia è previsto il trattamento con GH ricombinante per la sindrome di Turner e la sindrome di Prader-Willi (nota 39). La sindrome di Turner può presentare un fenotipo clinico piuttosto variabile, il cui unico segno può essere la bassa statura. Escluse le cause più frequenti di bassa statura, è opportuno eseguire nelle bambine l’esame del cariotipo.
Mutazione del gene SHOX (2): il gene risiede nel braccio corto dei cromosomi sessuali nella regione pseudo-autosomica. Sono state descritte diverse mutazioni a carico di questo gene, più frequentemente delezioni e mutazioni puntiformi, che possono determinare aspetti clinici variabili. È descritto un ampio spettro fenotipico, che va dalla bassa statura semplice (mutazioni di SHOX sono riscontrate nel 2-15% dei pazienti con bassa statura idiopatica), alla sindrome di Leri-Weill, alla sindrome di Langer. Nei bambini con mutazione del gene SHOX la lunghezza alla nascita è solo minimamente ridotta e dalla prima infanzia si assiste a una progressiva riduzione della velocità di crescita. Possono essere associate anomalie scheletriche caratteristiche, tra cui il segno di Madelung con sublussazione spontanea dell’ulna distale, mesomelia degli arti con riduzione dello span e della lunghezza delle gambe. Segni radiologici caratteristici, sebbene non sempre presenti, sono la triangolarizzazione, piramidalizzazione e “lucentezza” del radio distale. La diagnosi clinica viene confermata mediante indagine genetica. Nei pazienti con bassa statura dovuta a mutazione del gene SHOX è possibile la prescrizione della terapia con GH (nota 39).
Patologie scheletriche (osteocondrodisplasie): sono tipicamente caratterizzate da bassa statura disarmonica, con alterazioni nei rapporti antropometrici. Esistono più di cento forme diverse di displasie scheletriche. Tra le più note è sicuramente l’acondroplasia (3) (1:25.000 nati vivi), caratterizzata da rizomelia, facies caratteristica con bozze frontali prominenti e ipoplasia medio-facciale, lordosi lombare, ginocchio varo e macrocefalia. Tra le altre forme ricordiamo l’ipocondroplasia, la displasia spondilo-epifisaria, l’osteogenesi imperfetta.
SGA senza catch-up growth: si definisce SGA (small for gestational age) il bambino che presenta alla nascita peso o lunghezza < -2 DS rispetto alla media per sesso ed età gestazionale. Nel 90% circa dei casi, entro i primi due anni di vita si assiste al fenomeno del catch-up growth, caratterizzato da recupero staturo-ponderale. Soprattutto nei bambini nati pretermine tale recupero può avvenire anche più tardivamente, ma comunque entro i 4 anni di vita. Nel restante 10% dei casi non si ha recupero spontaneo ed è possibile, nel caso di statura ≤ - 2.5 DS e velocità di crescita < 50° centile, prescrivere terapia con GH dopo i 4 anni di età (nota 39). La risposta alla terapia con GH è comunque variabile e uno studio recente correla tali variazioni inter-individuali con diversi pattern genetici di secrezione e sensibilità insulinica (4). Spesso i bambini SGA presentano inoltre pubertà precoce, con ridotta ampiezza dello spurt puberale.
Malattie e infezioni croniche (malattie cardiache, renali, onco-ematologiche, metaboliche, polmonari e infezioni): qualunque malattia cronica severa può determinare rallentamento della crescita, sia per alterazioni legate alla patologia in sé, sia per le terapie che ne conseguono (1).
Malnutrizione (diminuito introito, malassorbimento): tra le cause di malassorbimento la più frequente è la celiachia, che può presentare come unico segno proprio la bassa statura. In ogni bambino con bassa statura deve essere eseguito lo screening per celiachia mediante dosaggio degli anticorpi anti-transglutaminasi e delle IgA totali (1).
Fattori psico-sociali
Cause iatrogene (farmaci, irradiazione): i glucocorticoidi rappresentano una delle cause iatrogene più frequenti di rallentamento della velocità di crescita. Questa categoria di farmaci agisce sulla crescita staturale attraverso diversi meccanismi, tra cui l’alterazione della secrezione e la modulazione dell’azione periferica del GH endogeno, e l’interferenza con il processo di neoformazione ossea e del collagene. Gli effetti sono variabili in funzione del tipo di farmaco, della dose e della durata della terapia e sono sostanzialmente reversibili alla sospensione del trattamento.
La radioterapia può essere causa di severi deficit staturali, soprattutto quando viene eseguita a livello cerebrale e della colonna. Il danno ipotalamo-ipofisario può manifestarsi anche tardivamente rispetto alla terapia e può determinare deficit pluritropinico, con rallentamento della velocità di crescita. L’irradiazione della colonna può determinare un alterato accrescimento del tronco, con alterazione dei rapporti tronco/arti.
La bassa statura idiopatica è una diagnosi di esclusione (5). La statura del bambino è al di sotto del range previsto per il target familiare, l’età ossea non è ritardata e devono essere state escluse le cause patologiche di bassa statura. Benchè possa essere considerata una variante “normale” di crescita, è importante controllare periodicamente il bambino per escludere cause patologiche sottostanti non riconosciute inizialmente.
BIBLIOGRAFIA
- Rogol AD, Hayden GF. Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr 2014, 164 (5 Suppl): S1-14.
- Binder G. Short Stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr 2011, 75: 81–9.
- Bellus GA, Hefferon TW, Ortiz de Luna RI, et al. Achondroplasia is defined by recurrent G380R mutations of FGFR3. Am J Hum Genet 1995, 56: 368-73.
- Beck Jensen R, Thankamony A, Day F, et al. Genetic markers of insulin sensitivity and insulin secretion are associated with spontaneous postnatal growth and response to growth hormone treatment in short SGA children: the North European SGA Study (NESGAS). J Clin Endocrinol Metab 2015, 100: E503-7.
- Cohen P, et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J Clin Endocrinol Metab 2008, 93: 4210–7.
Inquadramento diagnostico della crescita eccessiva
Sergio Bernasconi & Silvia Merli
Dipartimento di Medicina Clinica e Sperimentale, Università di Parma, Clinica Pediatrica, Azienda Ospedaliero-Universitaria di Parma
DEFINIZIONE
L’alta statura è generalmente definita come lunghezza o altezza ≥ 97° percentile rispetto alla popolazione di riferimento per sesso ed età, oppure maggiore di 2 DS sopra la mediana per una popolazione definita. Definizioni meno rigorose intendono lunghezza o altezza ≥ 95° percentile (z-score ≥ 1.66).
Sul piano clinico devono essere comunque indagati anche quei bambini con altezza normale ma superiore a 2 DS rispetto al target genetico (1).
CLASSIFICAZIONE
Dal punto di vista eziologico, le cause di iperaccrescimento/alta statura possono essere classificate come illustrato nella tabella 1.
| Tabella 1 Classificazione eziologica delle cause di alta statura |
|
| Varianti della norma | Alta statura familiare Avanzamento costituzionale di crescita |
| Eccesso o modulazione di fattori di crescita | IGF-I (esempio obesità) Insulina (esempio obesità) IGF-II (esempio, s. di Beckwith-Wiedemann) |
| Eccessiva produzione di steroidi sessuali | Pubertà precoce GnRH-dipendente Pubertà precoce GnRH-indipendente |
| Carenza di fattori necessari per arrestare la crescita lineare | Ipogonadismo ipogonadotropo Deficit di aromatasi Carenza di recettori estrogenici |
| Eccesso di geni dell’accrescimento | SHOX (s. di Klinefelter) Gene di controllo Y-specifico |
| Eccesso di secrezione di GH | Adenoma ipofisario GH-secernente S. di McCune-Albright MEN-1 Complesso di Carney |
| Eccesso o mutazione dei recettori dei fattori di crescita | Trisomia IGF-1R Inattivazione di FGFR3 |
Dal punto di vista clinico le cause di alta statura possono essere classificate valutando il periodo in cui tale caratteristica si manifesta (tabella 2): infatti, mentre un tumore GH-secernente può causare alta statura già nell'infanzia, disturbi che compromettono la produzione di steroidi sessuali o il loro metabolismo di solito si traducono in alta statura in adolescenza e nella prima età adulta, fase in cui gli steroidi sessuali sono variabili importanti nel processo di crescita (2,4).
| Tabella 2 Classificazione delle cause di alta statura secondo l’età di insorgenza |
||
| Epoca prenatale e prima infanzia | S. di Sotos S. di Beckwith-Wiedemann S. di Simpson-Golabi-Behmel S. di Perlman S. di Weaver Marshall S. di Pallister-Killian |
|
| Infanzia e adolescenza | Cause non endocrine | Alta statura familiare (costituzionale) Obesità esogena S. di Klinefelter S. di Marfan Omocistinuria |
| Cause endocrine | Pubertà precoce Eccesso di GH: adenoma ipofisario GH-secernente, s. di McCune-Albright, MEN-1, complesso di Carney Ipogonadismo Patologie coinvolgenti gli ormoni sessuali: deficit di aromatasi (10), carenza di recettori estrogenici Ipertiroidismo |
|
SINDROMI DA IPERACCRESCIMENTO CHE SI MANIFESTANO NEL PERIODO NEONATALE
Sindrome di Sotos
La sindrome di Sotos (gigantismo cerebrale) è una forma di gigantismo rara, con un'incidenza di 1 su 15000, caratterizzata da alcuni segni particolari, come la crescita eccessiva durante l'infanzia, la macrocefalia e le difficoltà di apprendimento di grado variabile.
La diagnosi solitamente viene sospettata dopo la nascita, per il significativo aumento del peso e della circonferenza cranica, l'ipotonia, le difficoltà all'alimentazione e le caratteristiche somatiche (viso lungo e stretto, mento appuntito, fronte bombata e larga, capelli sottili, ipertelorismo con rime palpebrali oblique verso il basso); caratteristiche meno comuni sono anomalie genito-urinarie, cardiache e crisi epilettiche. Il deficit cognitivo e il ritardo nello sviluppo motorio sono variabili. Il rischio riportato di tumori è del 2-4%
La maggior parte dei bambini affetti ha età ossea avanzata. Le mutazioni e le delezioni del gene NSD1 (localizzato sul cromosoma 5q35) sono responsabili del 60-90% dei casi. La secrezione di GH e le concentrazioni di IGF-I e ALS sono normali in questi soggetti (5).
Sindrome di Beckwith-Wiedemann
La sindrome deve essere sospettata in bambini con macrosomia, macroglossia, onfalocele/ernia ombelicale/diastasi dei muscoli retti dell'addome, visceromegalia. I bambini affetti sono alti e mostrano avanzamento dell'età ossea e rapida crescita durante la prima infanzia, con successivo rallentamento; l'altezza finale è al di sopra del range familiare. Altre caratteristiche cliniche comprendono ipoglicemia con iperinsulinismo, anomalie renali, emi-ipertrofia e rischio di sviluppare tumori embrionali (tumore di Wilms ed epatoblastoma).
La sindrome di Beckwith-Wiedemann è dovuta ad alterazioni genetiche e/o epigenetiche che alterano la funzione dei geni sottoposti a imprinting sul cromosoma 11p15.5. La malattia è sporadica nell'85% dei casi, mentre negli altri è presente trasmissione familiare. I diversi sottotipi molecolari comportano diversi rischi di ricorrenza e rischi clinici (ad esempio tumore). Nel 20% dei pazienti non sono state riscontrate anomalie molecolari.
La diagnosi generalmente è supportata dalla presenza di almeno tre segni clinici caratteristici.
Sindrome di Simpson-Golabi-Behmel
La sindrome è caratterizzata da iperaccrescimento pre- e post-natale, dismorfismi facciali (macrocefalia, ipertelorismo, mandibola prominente, macroglossia). In circa 1/3 dei casi sono presenti cardiopatie e possono essere presenti malformazioni viscerali e scheletriche varie e incostanti.
La prevalenza non è nota. Il rischio di sviluppare tumori embrionali è stimato del 7.5%. La trasmissione è recessiva legata all'X. Il gene principale è stato mappato in Xq26 e codifica per un proteoglicano extra-cellulare, GPC3, espresso soprattutto nei tessuti di derivazione mesodermica. Il GPC3 interagisce con IGF-II, interferendo con l’attività di segnale. La mutazione del gene GPC3 non è presente in tutti i pazienti; questo suggerisce che in alcuni casi possano essere implicati altri loci (6,7).
Altre rare sindromi associate ad eccessivo accrescimento includono la sindrome di Perlman, la sindrome di Weaver, Marshall e la sindrome di Pallister- Killian.
CRESCITA STATURALE ECCESSIVA NELL'INFANZIA E NELL'ADOLESCENZA
Può essere il risultato di fattori familiari, di disturbi endocrini e non endocrini e di condizioni sindromiche.
Cause non endocrine
Alta statura familiare (costituzionale)
Rappresenta il motivo più frequente di richiesta di valutazione per alta statura e solitamente è evidente dai 4 anni di età. Si verifica più frequentemente nelle femmine.
In tali soggetti sembra esserci una secrezione di GH e livelli di IGF-I relativamente più alti rispetto a bambini di media o bassa altezza. La valutazione clinica di tali soggetti risulta nella norma e la loro altezza generalmente si colloca nel range familiare, calcolato sulla base dell’altezza misurata nei genitori. Si può osservare in tali soggetti un aumento della velocità di crescita nella prima infanzia fino a circa 4 anni di età, che si normalizza in seguito. L’età ossea può essere normale o avanzata e può essere utilizzata per la valutazione della prognosi di crescita. Questi bambini e i loro genitori devono essere rassicurati sul fatto che il pattern di crescita è normale, anche se in alcuni casi può essere necessario comunque un periodo di monitoraggio della crescita.
Obesità esogena
I bambini obesi risultano spesso più alti durante l'infanzia, ma l'altezza finale non è di solito aumentata. Vi è una maggiore predisposizione allo sviluppo di adrenarca, pubertà precoce, iperinsulinismo ed età ossea avanzata, fattori che contribuiscono a un incremento della velocità di crescita e a una più alta statura durante l'infanzia.
Sindrome di Klinefelter
La sindrome 47,XXY è la più comune aneuploidia dei cromosomi sessuali causa di alta statura. La prevalenza è di circa 1-2/1000. Il fenotipo di tali soggetti è variabile, ma le caratteristiche cliniche comprendono alta statura, ginecomastia, difficoltà di apprendimento variabili e disturbi comportamentali, anomalie genitali (ipospadia, criptorchidismo). Tali soggetti solitamente raggiungono la pubertà spontaneamente, ma hanno ridotto volume testicolare con evidenza biochimica di insufficienza gonadica e conseguente infertilità.
Sindrome di Marfan
Si tratta di una malattia sistemica del tessuto connettivo, caratterizzata dall'associazione variabile di sintomi cardiovascolari, muscolo-scheletrici, oculari e polmonari. La prevalenza stimata è 1/5.000, senza differenza tra i sessi. Nella maggior parte dei casi, è causata dalle mutazioni del gene FBN1 (cromosoma 15q21) che codifica per la fibrillina-1, una proteina essenziale del tessuto connettivo. Alcune forme cliniche di confine sono dovute alle mutazioni del gene TGFBR2 (cromosoma 3), che codifica per un recettore del TGF-ß. La trasmissione è autosomica dominante.
Il coinvolgimento cardiovascolare è caratterizzato da: progressiva dilatazione dell'aorta (che può portare a insufficienza aortica), associata ad aumento del rischio di dissezione aortica, che compromette la prognosi; insufficienza mitralica, che può essere complicata da aritmie, endocarditi o insufficienza cardiaca.
Il coinvolgimento dello scheletro è spesso il primo segno della malattia e comprende dolico-stenomelia (eccessiva lunghezza delle estremità), sovrappeso, aracno-dattilia, ipermobilità delle articolazioni, scoliosi, protrusione dell'acetabolo, deformità del torace (torace carenato o pectus excavatum), dolicocefalia dell'asse antero-posteriore, micrognazia o ipoplasia della regione mascellare.
I segni oculari comprendono miopia assiale, che provoca distacco di retina, e dislocazione del cristallino (segni caratteristici sono l'ectopia o la lussazione). Le complicazioni oculari, soprattutto l'ectopia del cristallino, possono esitare nella cecità (8).
Omocistinuria
Si tratta di una patologia autosomica recessiva, dovuta alla mutazione del gene della cistationina ß-sintasi. I pazienti con omocistinuria hanno caratteristiche cliniche simili ai pazienti con sindrome di Marfan, ma con associati disturbi nell'apprendimento, maggiore incidenza di malattie psichiatriche e tendenza allo sviluppo di patologie trombo-emboliche. La diagnosi è confermata dal riscontro di elevati livelli di omocisteina plasmatica.
Cause endocrine
Pubertà precoce
Un precoce sviluppo sessuale, sia per cause centrali (gonadotropino-dipendenti, idiopatica o secondaria ad anomalie del sistema nervoso centrale) che periferiche (gonadotropino-indipendenti, da tumori testicolari e ovarici, cisti ovariche, sindrome di McCune-Albright, tumori hCG-secernenti), può accelerare la crescita staturale, con marcato avanzamento della maturazione ossea. L’azione degli ormoni sessuali provoca infatti maturazione dei condrociti e crescita ossea longitudinale. Questo provoca una precoce fusione delle epifisi ossee, con compromissione dell’altezza finale.
Eccesso di produzione di ormone della crescita
Un’eccessiva produzione di GH in età pediatrica è nella maggior parte dei casi causata dalla presenza di un adenoma ipofisario GH-secernente. Il gigantismo ipofisario in genere è una condizione sporadica e isolata. Tuttavia, si può verificare nel contesto di una condizione sindromica o secondo uno schema di trasmissione familiare. Sindromi in cui il gigantismo è una caratteristica ben riconosciuta includono la sindrome di McCune-Albright, la MEN-1 e il complesso di Carney.
La diagnosi si basa sull’esecuzione di test di soppressione e sul dosaggio dei livelli di IGF-I. Di fondamentale importanza è l’esecuzione nei casi sospetti di RMN encefalo con studio ipofisario.
Deficit o resistenza agli ormoni sessuali
Una condizione di ipogonadismo permanente provoca ritardata maturazione scheletrica, con prolungato periodo di accrescimento e alta statura. Ad esempio, sono stati descritti pazienti con deficit permanente di estrogeni, risultante da un deficit di aromatasi o da una resistenza agli estrogeni dovuta a mutazione del gene del recettore degli estrogeni. In questi soggetti si verifica incompleta chiusura delle epifisi con alta statura, associata anche ad osteopenia (10).
Ipertiroidismo
L’ipertiroidismo da iperproduzione endogena di ormoni tiroidei o sovradosaggio con ormoni esogeni può provocare un incremento della crescita e avanzamento dell’età ossea. Il trattamento, ristabilendo una condizione di eutiroidismo, normalizza il ritmo di crescita.
APPROCCIO DIAGNOSTICO
L’approccio diagnostico nei confronti di un bambino con iperaccrescimento/alta statura può essere sintetizzato come in figura 1.

Figura 1
Di fondamentale importanza nell’approccio al bambino con alta statura è un’anamnesi accurata, a partire da peso e lunghezza alla nascita, storia della gravidanza ed eventuali comorbilità, nonché la valutazione dell’altezza dei genitori, per il calcolo del target genetico
La valutazione clinica deve comprendere la rilevazione dei parametri auxologici, dello stadio puberale e della presenza di eventuali dimorfismi o caratteri sindromici, in modo da orientare la diagnosi differenziale delle diverse cause di alta statura e guidare i successivi approfondimenti diagnostici. La valutazione auxologica deve prevedere, oltre alla valutazione di peso e altezza, da riportare sugli specifici percentili per sesso ed età, il calcolo del BMI. Nel determinare il trend di crescita in età pediatrica, rispetto a una singola misurazione, risulta maggiormente significativa la determinazione della velocità di crescita; ciò richiede misurazioni seriate dell'altezza, a distanza di almeno 4-6 mesi le une dalle altre, preferibilmente ad opera dello stesso operatore e con la stessa strumentazione.
Altri parametri da valutare al momento della visita auxologica, che possono essere di fondamentale importanza nella diagnosi differenziale tra le diverse cause, sono la misura dell’altezza da seduto (sitting height) per valutare la presenza di alterate proporzioni tra i segmenti corporei (es. sindrome di Marfan, trisomia X), la misura dell’ apertura delle braccia (superiore all’altezza nelle s. di Marfan e di Sotos) e della circonferenza cranica (> 98° centile nella maggior parte dei pazienti con s. di Sotos). È inoltre essenziale una valutazione degli stadi puberali per escludere la presenza di pubertà precoce.
La scelta delle indagini da eseguire nei casi di iperaccrescimento dipende dal tipo di sospetto clinico e dalla presenza di eventuali segni e sintomi caratteristici. Di fondamentale importanza è quindi l’esecuzione di esami ematici (FT4 e TSH), la valutazione dell’asse GH-IGF-I (dosaggi basali e, quando necessario, esecuzione di test di soppressione) e lo studio dell’età ossea attraverso l’esecuzione di Rx del polso e della mano sinistra. Nei soggetti con un'anomalia di sviluppo puberale è necessario il dosaggio di LH, FSH, estradiolo e testosterone ed eventualmente, nei casi di pubertà precoce, la valutazione di alfa-feto-proteina e hCG.
In caso di dismorfismi, ritardo mentale e/o altre caratteristiche sindromiche, si consiglia l’esecuzione di consulenza genetica e l’eventuale esecuzione di cariotipo. In presenza di caratteristiche marfanoidi, è da prendere in considerazione l’esecuzione di dosaggio di omocisteinemia e ricerca delle mutazioni FBN1 (2,3,9).
BIBLIOGRAFIA
- Neylon OM, et al. Overgrowth syndromes. Curr Opin Pediatr 2012, 24: 505-11.
- Kumar S. Tall stature in children: differential diagnosis and management. Int J Pediatr Endocrinol 2013, 2013 (suppl 1): P53.
- Davies JD, Cheetham T. Investigation and management of tall stature. Arch Dis Child 2014, 99: 772-7.
- Sabin MA, et al. Genetics of obesity and overgrowth syndromes. Best Pract Res Clin Endocrinol Metab 2011, 25: 207-20.
- Tatton-Brown K, et al. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet 2005, 77: 193-204.
- Mariani S, Iughetti L, Bertorelli R, et al. Genotype/phenotype correlations of males affected by Simpson-Golabi-Behmel syndrome with GPC3 gene mutations: patient report and review of the literature. J Pediatr Endocrinol Metab 2003, 16: 225-32.
- Cottereau E, et al. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet 2013, 163C: 92-105.
- Bread T, et al. Health supervision for children with Marfan syndrome. Am Acad Pediatr 2009, 123: 1556-61.
- Moon RJ, Davies JH. Evaluation of tall stature. Paediatr Child Health 2010, 20: 43-5.
- Rochira V, et al. Tall stature without growth hormone: four male patients with aromatase deficiency. J Clin Endocrinol Metab 2010, 95: 1626–33.
Approccio terapeutico alla crescita eccessiva
Sergio Bernasconi & Silvia Merli
Dipartimento di Medicina Clinica e Sperimentale, Università di Parma, Clinica Pediatrica, Azienda Ospedaliero-Universitaria di Parma
L’approccio terapeutico può ovviamente variare in base alla causa che ha determinato l’alta statura.
Un eccesso, per esempio, di secrezione di GH richiederà un intervento di adenomectomia dell’ipofisi e/o una terapia farmacologica, con analoghi della somatostatina, pegvisomant o in casi più lieve cabergolina (1).
Molto discussa è la terapia farmacologica nelle alte stature costituzionali isolate o familiari, in cui l’indicazione terapeutica non è legata a una patologia, ma può trovare un razionale in aspetti psicologici, sociali-relazionali ed estetici. Vi è da sottolineare che questa terapia veniva maggiormente presa in considerazione in passato, soprattutto nel sesso femminile, quando probabilmente era minore il grado di accettazione sociale di persone molto alte (2).
Nei maschi è stato utilizzato il testosterone enantato, a dosi variabili da 250 a 500 mg ogni due settimane per 12-18 mesi. Con la dose inferiore si ottiene lo stesso risultato di diminuzione della statura finale di circa 9 cm ed è quindi preferibile. Il follow-up a lunga distanza, in età adulta, dei soggetti trattati in adolescenza ha dimostrato un normale tasso di paternità e una normale qualità del seme, anche se i livelli ematici di testosterone tendono ad essere più bassi rispetto ai non trattati (3).
Ridotta fertilità è stata invece descritta in donne che erano state trattate durante l’adolescenza con 200 µg/die di etinil-estradiolo, mentre questo dato non è così evidente in quelle che hanno assunto una dose inferiore (100 µg/die). Vi è anche da sottolineare che le dosi impiegate da alcuni autori sono state anche di 500-1000 µg/die. La diminuzione di statura finale è stata in media di 5-7 cm (4, 5).
In conclusione, la terapia con androgeni ed estrogeni per le alte stature non appare oggi proponibile ed è da considerarsi frutto di convinzioni sociali ormai superate. Aperto è invece il dibattito sul possibile uso di questi farmaci per ridurre l’altezza in bambini con gravi disabilità cognitive (6). È questo un tema di estrema difficoltà da affrontare per i riflessi psicologici, sociali, etici che comporta. Le società scientifiche dovrebbero porvi maggiore attenzione, cercando anche di fissare alcune linee generali di comportamento che possano aiutare il singolo clinico a non dover affrontare da solo una problematica così vasta.
Bibliografia
- Andersen M. GH excess: diagnosis and medical therapy. Eur J Endocrinol 2013, 170: R31-41.
- Lee JM, Howell JD. Tall girls: the social shaping of a medical therapy. Arch Pediatr Adolesc Med 2006, 160: 1035-9.
- Reinehr T, Gueldensupp M, Wush R, et al. Treatment of tall stature in boys: comparison of two different treatment regimens. Horm Res Paediatr 2011, 76: 343-7.
- Hendriks AE, Boellaard WP, van Casteren NJ, et al. Fatherhood in tall men treated with high-dose sex steroids during adolescence. J Clin Endocrinol Metab 2010, 95: 5233-40.
- Hendriks AE, Drop SL, Laven JS, et al. Fertility of tall girls treated with high-dose estrogen, a dose-response relationship. J Clin Endocrinol Metab 2012, 97: 3107–14.
- Allen DB, et al. Growth-attenuation therapy: principles for practice. Pediatrics 2009, 123: 1556-61.
Differenze dello sviluppo del sesso
Questa sezione è pubblicata grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Fisiologia dello sviluppo dell'apparato riproduttivo:
Classificazione e nuova nomenclatura
- DSD da alterazione nel numero dei cromosomi sessuali
- DSD 46 XY
- DSD 46 XX
- DSD con ovotestis
- DSD di difficile classificazione
Protocollo diagnostico e management in età neonatale
Attribuzione del genere e correzione delle ambiguità genitali
Introduzione e overview sui DSD
Antonio Balsamo1, Silvano Bertelloni2, Franco D’Alberton1, Giacinto Marrocco3, Santiago Vallasciani4
1Università di Bologna, AOU Policlinico S.Orsola-Malpighi; 2UO Pediatria 1, AOU Pisa; 3Chirurgia e Urologia pediatrica, Ospedale San Camillo, Roma; 4Urologia Pediatrica, Ospedale Maggiore Policlinico Mangiagalli e Regina Elena, Milano
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
Negli esseri umani, la diversificazione sessuale è associata alla presenza di 2 cromosomi XX nelle femmine, e di un cromosoma X e uno Y nei maschi. I cromosomi X e Y hanno caratteristiche e corredo genetico diversi tra loro (circa 1.000 geni sulla X e solo poche decine di geni sull’Y). Una serie di ricombinazioni seguita da perdita di materiale genetico sul gene Y ha portato alla differenziazione morfologica dei 2 cromosomi sessuali (1). La maggior parte dei geni sui cromosomi sessuali non sono direttamente coinvolti nella determinazione sessuale, sebbene lo sviluppo di un feto in senso maschile per lungo tempo sia stato attribuito principalmente alla presenza del singolo gene SRY sul cromosoma Y, maschio-limitante. In realtà studi più recenti hanno dimostrato che questo caposaldo è molto più “variabile” di quanto ipotizzato e meccanismi alternativi possono innescare uno sviluppo sessuale diverso da quello atteso in base al solo cariotipo determinatosi all’atto della fecondazione. Oggi, l'idea che lo sviluppo in senso femminile sia un'opzione predefinita passiva (sesso di default) è stata accantonata dopo la scoperta dei geni che promuovono attivamente lo sviluppo delle ovaie e sopprimono il programma testicolare. Un articolo pubblicato nel 2015 (2) illustra lo spettro di differenze che possono essere presenti in questo aspetto dello sviluppo umano (tabella).
| Tabella 1 Spettro della variabilità del sesso cromosomico, gonadico, fenotipico nell'uomo (mod da 2) |
|||||||||
| Fenotipo | Maschio tipico | Differenze lievi | Differenze moderate | 46,XY DSD | DSD ovo-testicolare | 46,XX DSD testicolare | Moderate variazioni | Sottili variazioni | Femmina tipica |
| Cromosomi | XY | XY | XY | XY | XY, XX o un misto di entrambi | XX | XX | XX | XX |
| Gonadi | Testicoli | Testicoli | Testicoli | Testicoli | Tessuto sia ovarico che testicolare | Piccoli testicoli | Ovaie | Ovaie | Ovaie |
| Genitali | Interni ed esterni maschili | Interni ed esterni maschili | Esterni maschili, ipospadia | Spesso “ambigui” | "Ambigui" | Esterni maschili | Esterni maschili e interni femminili | Interni e esterni femminili | Interni e esterni femminili |
| Altre caratte-ristiche/ esempi | Caratte-ristiche sessuali secondarie maschili | Oligo-spermia, bassa fertilità | 1:250-1:400 neonati | La sindrome da “persistenza dei dotti di Muller” presenta testicoli e genitali esterni maschili, ma anche utero e tube | Rare segnalazioni di persone XY che concepiscono e crescono un figlio sano | Causato generalmente dalla presenza del gene SRY | Eccesso di ormoni sessuali maschili | Insufficienza ovarica precoce o ovaio policistico | Caratteristiche sessuali secondarie femminili |
Pertanto, la determinazione del sesso a livello molecolare, gonadico o fenotipico può essere molto più complicata rispetto alla classica visione binaria maschio/femmina del passato. Classicamente, si riteneva, infatti, che la presenza o l'assenza di un cromosoma Y fosse l’aspetto sufficiente e necessario: “con esso si è maschio, e senza di esso si è “femmine”. Al contrario, chi si occupa di “differenze dello sviluppo del sesso” sa che alcune persone si trovano “a cavallo”, nel senso che i loro cromosomi sessuali affermano una cosa, ma le loro gonadi (ovaie o testicoli) o le loro caratteristiche fenotipiche ne mostrano un’altra.
Quando entra in campo la genetica, il confine tra i sessi diventa ancora più sfuocato. Infatti, gli studiosi hanno identificato un numero sempre maggiore di geni coinvolti nelle principali forme di DSD (acronimo Inglese per “Differences of Sex Development”), e la loro scoperta ha permesso di comprendere da un lato la patogenesi di forme precedentemente senza diagnosi e dall’altro che variazioni di questi geni possono avere effetti sottili sul sesso anatomico o fisiologico della persona.
L’evidenza che esistessero geni per fattori di trascrizione sesso-determinanti e molecole-segnale è emersa inizialmente dall’identificazione di anomalie cromosomiche e successivamente dall’identificazione di mutazioni in geni come SRY, SOX9, NR5A1, WT1, DAX1, WNT4, CBX2, DMRT1 e GATA4 in persone con DSD gonadico (3). Da questi studi è poi derivata l’acquisizione che la dose del gene e il livello di espressione genetica risultante può essere critica per la determinazione testicolare. L’espressione di una singola copia dei geni SOX9, SF1 e WT1, così come duplicazioni dei geni DAX1 e WNT4 in persone con cariotipo 46,XY possono condurre a disgenesia gonadica, mentre duplicazioni dei geni SOX9 o SOX3 possono portare a 46,XX DSD testicolare (3). Più recenti studi hanno, infine, messo in evidenza che specifiche mutazioni attivanti nel gene trasduttore di segnali, MAP3K1, causano disgenesia gonadica parziale o completa, alterando le attività di molecole segnale e fattori di trascrizione, compresi β-catenina e SOX9.
Molti di questi nuovi geni sono stati identificati mediante l’applicazione di moderne tecnologie, comprese l’ibridizzazione genomica comparativa (CGH array) e il sequenziamento del genoma. Insieme, questi recenti sviluppi forniscono una visione più ampia del controllo genetico della determinazione del sesso e delle sue “differenze”. Le nuove tecnologie nel sequenziamento del DNA e la biologia cellulare stanno però rivoluzionando molti concetti, suggerendo che ognuno di noi è, a vari livelli, un mosaico di cellule geneticamente distinte, alcune con un sesso che potrebbe non corrispondere a quello del resto del loro corpo. Alcuni studi suggeriscono che il sesso di ogni cellula possa poi influenzare il comportamento generale dell’individuo, attraverso una rete complessa di interazioni molecolari. L’evoluzione delle metodiche di studio del genoma consentirà di ampliare ulteriormente lo spettro di condizioni note e auspicabilmente renderà più motivato e personalizzato ogni intervento in questo campo.
Queste scoperte mal si conciliano con un mondo in cui il sesso è ancora definito in termini binari. Pochi sistemi giuridici consentono una qualsiasi ambiguità del sesso biologico, mentre i diritti legali di una persona e il suo status sociale possono essere fortemente influenzati se il suo certificato di nascita dice maschio o femmina. Mentre la salute generale e le capacità cognitive delle persone con DSD sono solitamente normali, la diagnosi può risultare difficile per essi stessi e i loro genitori (4). Gli individui potrebbero essere assegnati a un sesso di crescita discordante dalla propria identità di genere e talvolta ciò può condurre a una successiva insoddisfazione di genere e potrebbe richiedere una riassegnazione. La persona con DSD può aver bisogno di una chirurgia correttiva dei genitali esterni e interni, della rimozione parziale o completa delle gonadi disgenetiche o di un ovo-testis, dell’eventuale riposizionamento delle gonadi e di una terapia ormonale sostitutiva (TOS) nell’infanzia o nell’adolescenza, da continuare fino all’età adulta. Inoltre, la persona con DSD potrebbe non essere l’unico membro portatore della condizione nella famiglia.
Nei diversi capitoli di questa sezione verranno descritte, oltre le più comuni cause di DSD, i più recenti sviluppi nella genetica di queste condizioni e la multidisciplinarietà indispensabile per la loro presa in carico adeguata.
Bibliografia
- Charlesworth B. The evolution of chromosomal sex determination and dosage compensation. Curr Biol 1996, 6: 149–62.
- Ainsworth BYC. Sex redefined. Nature 2015, 518: 288–91.
- Ostrer H. Disorders of sex development (DSDs): an update. J Clin Endocrinol Metab 2014, 99: 1503–9.
- Schober J, Nordenström A, Hoebeke P, et al. Disorders of sex development: summaries of long-term outcome studies. J Pediatr Urol 2012, 8: 616–23.
Determinazione e differenziazione sessuale
Paola Grammatico, Silvia Majore
Genetica, Ospedale San Camillo-Forlanini, Università Sapienza, Roma
(questo capitolo è pubblicato grazie a un accordo con il Gruppo di Studio Italiano DSD, www.gruppodistudio-it-dsd.org che detiene il copyright di tutti i paragrafi contrassegnati con il seguente logo “copyright ![]() tutti i diritti sono riservati”)
tutti i diritti sono riservati”)
INTRODUZIONE
Lo “sviluppo sessuale” definisce l’insieme degli eventi biologici che conducono all’acquisizione differenziale, morfologica e funzionale, dei caratteri sessuali negli individui di sesso femminile e maschile, prerogativa necessaria a garantire la riproduzione e, con questa, il mantenimento della specie. Si realizza attraverso complesse reti di segnali sequenziali geneticamente determinati, dose- e tempo-dipendenti, che nell’uomo differiscono nei due sessi sin dalla VII settimana dello sviluppo embrionale, e termina alla pubertà con l’acquisizione della capacità riproduttiva (1,2).
Nelle prime fasi dello sviluppo embrionale, prima che la differenziazione delle gonadi diverga in senso maschile o femminile, ha luogo, in entrambi i sessi, la formazione di strutture primordiali bipotenti a controllo genico sesso-indipendente (3).
Lo sviluppo sessuale dimorfico comprende due processi, distinti ma sequenziali (4,5):
- la “determinazione sessuale” indirizza la differenziazione sesso-specifica della gonade in relazione al sesso cromosomico stabilitosi al momento della fecondazione;
- la “differenziazione sessuale”, che dipende dalla presenza o meno del testicolo nella fase dello sviluppo intra-uterino (differenziazione primaria), comprende la formazione dei dotti genitali e dei genitali esterni e, in seguito, le modificazioni sesso-specifiche che si verificano alla pubertà.
SVILUPPO DELLA GONADE INDIFFERENZIATA
Le gonadi si formano, in entrambi sessi, nell’ambito del sistema uro-genitale in via di sviluppo. Quest’ultimo, a sua volta, origina dal mesoderma intermedio, da cui si differenziano in sequenza cranio-caudale il pronefro, il mesonefro e il metanefro. La morfogenesi del primordio gonadico bipotente ha inizio in entrambi i sessi intorno alla IV settimana di gestazione, quando compare la cresta genitale, struttura pari costituita da un ispessimento della regione mediale del mesonefro e dalla proliferazione dell’epitelio celomatico sovrastante.
Il primordio gonadico, morfologicamente identico nei due sessi, è costituito da cellule celomatiche che formano uno strato superficiale (cortex) e da una regione interna, detta medulla. Quest’ultima contiene cellule provenienti dal mesonefro e cellule dell’epitelio celomatico, che, grazie alla frammentazione della membrana basale, invadono la medulla e si organizzano in cordoni compatti (cordoni sessuali primitivi). Infine, nell’ambito del primordio gonadico compaiono le cellule germinali primordiali (protogoni), migrate dalla parete del sacco vitellino verso la seconda metà della V settimana di gestazione (6).
Questa prima fase dello sviluppo gonadico si realizza grazie all’azione di molteplici geni, alcuni dei quali partecipano alla formazione di più organi, derivati o meno dal mesoderma intermedio. Tra questi, è stato a oggi attribuito un ruolo primario a (7):
- GATA4, che codifica per un fattore di trascrizione essenziale per l’iniziale sviluppo di molti organi, tra cui il cuore, è il gene che probabilmente dà inizio alla formazione della cresta gonadica;
- EMX2 è coinvolto nella formazione del sistema nervoso centrale e dell’intero sistema uro-genitale;
- WT1 è un gene la cui azione è essenziale nelle prime fasi della nefrogenesi e che interviene attivamente nello sviluppo del testicolo, oltre che del primordio gonadico;
- LHX9 svolge il suo ruolo a monte di SF1, gene inizialmente attivo nei primordi gonadici e surrenalici di entrambi i sessi e che in seguito, nel corso della formazione della gonade maschile e femminile, si esprime con modalità dimorfica;
- CBX2 è anch’esso un gene correlato allo sviluppo del primordio gonadico, ma che si esprime anche nelle fasi successive della differenziazione gonadica.

Figura 1. Sviluppo sessuale nel maschio e nella femmina. A: vengono indicati i principali geni coinvolti nella formazione del primordio gonadico bipotente. B: la determinazione sessuale è avviata nel maschio dal gene SRY, il cui principale bersaglio è SOX9. Questo, a sua volta, attiva l’espressione di molteplici geni target. In assenza di SRY e in presenza di 2 cromosomi X, l’azione di più geni determina lo sviluppo dell’ovaio. C: i fattori prodotti dal testicolo promuovono nel maschio lo sviluppo dei dotti di Wolff e dei genitali esterni e la degenerazione dei dotti di Müller. In assenza di ormoni maschili, in un ambiente estrogenizzato avviene la regressione dei dotti di Wolff e lo sviluppo differenziato dei dotti di Müller e dei genitali esterni femminili. Il gene WNT4 e altri fattori sono coinvolti nello sviluppo dei dotti genitali femminili.
DETERMINAZIONE SESSUALE
La determinazione sessuale si riferisce a quel processo per cui lo stesso organo primordiale viene indotto, a seconda dell’assetto dei cromosomi sessuali, a differenziarsi in testicolo o ovaio. Nei mammiferi lo sviluppo differenziale delle gonadi è controllato da numerosi geni, la cui espressione, “dose- e tempo-specifica”, promuove lo sviluppo di un sesso gonadico e/o antagonizza quello opposto. La successiva differenziazione sesso-specifica, che porta all’acquisizione fenotipica e funzionale dei caratteri maschili o femminili, dipende dalla corretta realizzazione di questo primo processo.
Determinazione testicolare
È noto da tempo (8,9) che SRY, localizzato nel braccio corto del cromosoma Y, rappresenta il gene della determinazione del sesso maschile. Infatti, la sua attivazione, ristretta a un preciso spazio temporale e dose-dipendente, è in grado di avviare, nella cresta genitale di embrioni 46,XY, un peculiare pattern a cascata di espressione genica, che coinvolge molti fattori di trascrizione e che produce la progressiva differenziazione morfologica e funzionale del primordio gonadico in testicolo e, allo stesso tempo, inibisce lo sviluppo della gonade femminile. Al contrario, in assenza di SRY e in presenza di due cromosomi X, prendono il sopravvento i pattern molecolari femminili, consentendo lo sviluppo dell’ovaio. A partire dalla VI settimana dalla fecondazione nell’embrione XY, poco dopo l’inizio dell’attivazione del gene SRY, si compie rapidamente la progressiva differenziazione della gonade primordiale bipotente in testicolo (2). Tale processo, a differenza di quanto avviene nella differenziazione ovarica, non viene alterato dall’eventuale assenza dei protogoni, che, quando fisiologicamente presenti, nella gonade determinata in senso maschile migrano dal cortex nei cordoni sessuali primari della medulla. Morfologicamente, la differenziazione del primordio gonadico in testicolo è innanzitutto caratterizzata da un significativo aumento di dimensioni dei cordoni sessuali primari, i quali si organizzano in strutture che costituiscono l’abbozzo dei tubuli seminiferi. Nel contesto di tali cordoni le cellule epiteliali si differenziano progressivamente in cellule del Sertoli. L’interstizio tra i cordoni viene invece invaso da vari tipi cellulari provenienti dal mesonefro, quali cellule mioidi che circondano i cordoni sessuali, cellule endoteliali e cellule mio-epiteliali, che nell’insieme formano lo stroma testicolare. Cellule mesenchimali della gonade primordiale si differenziano a loro volta in cellule del Leydig. Le cellule del Sertoli, individuabili prima delle cellule del Leydig, agiscono come centro organizzatore della gonade maschile, producono varie sostanze e, tra queste, l’ormone anti-Mülleriano (AMH) e l’inibina B. Successivamente (IX-X settimana), le cellule steroidogeniche del Leydig producono a loro volta steroidi androgeni e Insulin Like Factor 3 (INSL3) (10). Altri eventi fondamentali nell’organo-genesi testicolare sono la formazione della rete testis, a partire dalla porzione più interna dei cordoni sessuali, e la differenziazione dei duttuli efferenti, derivati dai tubuli mesonefrici (che connetteranno la rete testis ai derivati del dotto mesonefrico). Infine, i cordoni sessuali perdono il loro contatto con il cortex, tramite la formazione di un denso strato connettivo, denominato tunica albuginea.

Figura 2. Segnali opposti regolano la determinazione del sesso nel primordio gonadico o bipotente XY e XX. A: nel maschio SRY agisce da interruttore genetico e dà inizio al pathway differenziativo in direzione maschile, creando uno sbilanciamento fra segnali opposti, mediato dall’attivazione di SOX9. SOX9 upregola positivamente FGF9, il quale mantiene a sua volta l’espressione di SOX9, formando un circuito positivo nelle gonadi XY. In aggiunta FGF9 inibisce WNT4, gene coinvolto nella formazione dell’ovaio. Diversi segnali molecolari, tra cui il prodotto di SRY, reprimono l’espressione di RSPO1, uno degli induttori dello sviluppo ovarico. CYP26B1 si oppone all’inizio della meiosi mediata dall’acido retinoico, determinandone la degradazione. B: nella determinazione ovarica, verosimilmente non indotta da un unico gene, l’assenza di un segnale positivo per il circuito fra SOX9 e FGF9 permette l’iper-espressione di WNT4 e del suo gene target CTNNB1. WNT4 reprime FGF9, permettendo quindi l’attivazione di pattern molecolari di tipo femminile. L’inibizione dello sviluppo testicolare è anche mediata dall’azione di FOXL2, che reprime SOX9 e SF1. La mancata espressione di CYP26B1, in grado di degradare l’acido retinoico, rende possibile l’entrata in meiosi delle cellule germinali dell’ovaio durante lo sviluppo intra-uterino.
Le vie molecolari che regolano la differenziazione testicolare sono state in buona parte messe in luce e comprendono, dopo l’attivazione del gene SRY e il raggiungimento di una soglia critica della sua proteina nel corso di una stretta finestra temporale, l’attivazione di un articolato pattern genico specifico per il sesso maschile. Il principale bersaglio di SRY è oggi ritenuto essere SOX9 (17q24.3-q25.1), gene che gioca un ruolo chiave nello sviluppo testicolare e viene espresso con modalità distintamente dimorfica nel corso dell’intera gonado-genesi sesso-specifica. La sintesi della proteina SOX9, espressa a bassi livelli nel primordio gonadico bipotente, aumenta considerevolmente nella gonade XY poco dopo l’attivazione di SRY e si mantiene elevata anche dopo il breve periodo in cui agisce quest’ultimo gene. Nella gonade XX, al contrario, l’espressione di SOX9 si esaurisce con l’inizio della determinazione ovarica, giacché regolata negativamente da molteplici geni, compreso DAX1, che promuovono il differenziamento ovarico. A sua volta, la proteina SOX9, prodotta prevalentemente nelle cellule del Sertoli, dà inizio all’espressione di geni che codificano per molecole segnale attive a livello testicolare, quali FGF9, FGFR2, AMH, VNN, PGDS, CBLN4 e DHH, e regola in senso maschile quella di alcuni dei geni già coinvolti nello sviluppo del primordio gonadico, come ad esempio WT1 e SF1 (7,11). Il compimento della differenziazione testicolare viene reso possibile anche dal fatto che nella gonade specificata in senso maschile si stabiliscono circuiti a feed-back positivo (es. tra SOX9 e FGF9) che mantengono l’espressione di SOX9, e quindi dei suoi numerosi geni target, e inibiscono quella di geni coinvolti nello sviluppo dell’ovaio (es WNT4).
Determinazione ovarica
L’inizio dello sviluppo delle ovaie dal primordio gonadico bipotente ha inizio una settimana più tardi rispetto ai testicoli. È un processo notevolmente più lento, che si realizza solo in presenza dei protogoni, necessari per la formazione dell’unità anatomico-funzionale dell’ovaio stesso, ovvero il follicolo. Gli eventi morfologici che segnano la determinazione in senso ovarico della gonade primordiale sono, innanzitutto, la netta diminuzione delle dimensioni della medulla, nell’ambito della quale i cordoni sessuali primari si disgregano e vengono sostituiti da stroma vascolare. Al contrario, a livello del cortex le cellule dell’epitelio celomatico continuano a proliferare e si organizzano in cordoni sessuali secondari (corticali), che in seguito circondano i protogoni. Questi ultimi, a differenza di quanto avviene nella testicolo-genesi, tendono a non migrare nella medulla e sono inizialmente raggruppati in aggregati. In aggiunta, nelle fasi precoci della morfogenesi ovarica le cellule germinali mostrano una vivace attività mitotica (ovogoni). Verso la fine del III mese gli ovogoni vengono circondati da cellule epiteliali derivate dai cordoni sessuali secondari, che progressivamente si differenziano in cellule della granulosa (ritenute l’equivalente femminile delle cellule del Sertoli). Si costituiscono quindi i follicoli primordiali, strutture inizialmente composte da uno o più ovogoni uniti tra loro da ponti citoplasmatici e da un singolo strato di cellule della granulosa in via di differenziazione.
A circa 24 settimane dopo la fertilizzazione, nelle cellule germinali ha inizio la prima divisione meiotica, che si blocca allo stadio di diplotene. Gli ovogoni si differenziano quindi in ovociti di prim’ordine, il cui destino è, nella maggior parte dei casi, la degenerazione apoptotica. I follicoli residui, all’interno dei quali tende man mano a essere presente un singolo oocita, entrano invece in uno stato di quiescenza, che perdura fino alla pubertà.
A partire da questa fase della vita post-natale, in occasione di ciascun ciclo ovulatorio lo sviluppo di alcuni follicoli progredisce attraverso successivi stadi e si completa in una singola unità funzionale, nell’ambito della quale l’ovocita porta a termine la prima divisione meiotica.
Anche la differenziazione ovarica viene attivamente regolata da molteplici segnali molecolari: l'espressione di alcuni geni favorisce lo sviluppo ovarico e allo stesso tempo inibisce la comparsa di componenti proprie della gonade maschile (12). Non sembrerebbe però esistere, come nel testicolo, un singolo gene in grado d’innescare una specifica cascata ovaio-specifica. Le più recenti acquisizioni suggeriscono che l’espressione dei geni convolti nella differenziazione ovarica si esplichi, invece, perlopiù parallelamente e attraverso circuiti indipendenti, che permettono ciascuno il realizzarsi di un diverso aspetto del suo sviluppo (11,13). Le conoscenze dei geni coinvolti nella determinazione e nella differenziazione della gonade femminile sono comunque ancora piuttosto limitate e frammentarie. È degno di nota il fatto che alcuni dei geni espressi a elevati livelli durante lo sviluppo ovarico non rimangono inattivi durante la testicolo-genesi. Ciò va a sostegno del concetto che la determinazione del sesso si realizza grazie a un raffinato e complesso controllo nella regolazione quantitativa oltre che qualitativa dell’espressione genica. Il primo gene che risulta attivato all’inizio della gonado-genesi femminile è DAZL, il cui tradotto è verosimilmente coinvolto nell’induzione della meiosi negli ovogoni, quest’ultima principalmente mediata dall’acido retinoico (14). FOXL2 è uno dei geni maggiormente espressi nelle gonadi XX e sembra esercitare un ruolo fondamentale nel promuovere lo sviluppo dell’ovaio e nel reprimere la formazione di elementi propri del testicolo, grazie a un meccanismo di antagonismo reciproco, che viene mantenuto nel corso di tutta la vita. Un importante ruolo nella formazione dell’ovaio è stato inoltre attribuito ai geni WNT4, RSPO1 e CTNNB1, coinvolti in una cascata di segnali che produce l’attivazione della β-catenina (codificata da CTNNB1) e la conseguente regolazione positiva o negativa di una varietà di altri geni, molti ancora da identificare (15,16).
DIFFERENZIAZIONE SESSUALE
Con il processo di differenziazione sessuale si realizza lo sviluppo delle caratteristiche fenotipiche che contraddistinguono il sesso maschile e quello femminile. La sostanziale differenza nei due sessi risiede nel fatto che la fase intra-uterina della differenziazione del fenotipo sessuale, ovvero la morfogenesi degli ovidotti e dei genitali esterni, può realizzarsi, per il sesso maschile, solo a seguito della corretta formazione di un testicolo funzionante. Infatti, durante lo sviluppo embrionale il testicolo produce molteplici fattori, che dall’VIII settimana dirigono il successivo sviluppo dell’apparato riproduttivo in senso maschile. In mancanza di un testicolo funzionante, indipendentemente da quale sia l’assetto dei cromosomi sessuali e finanche in assenza dell’ovaio, la differenziazione dell’apparato genitale avviene in senso femminile. Entrambi i tipi di gonade e i loro prodotti ormonali sono invece necessari, durante la pubertà, per lo sviluppo dei caratteri sessuali secondari e per la funzionalità riproduttiva.
Sviluppo dei dotti genitali
Al pari di quanto avviene nelle gonadi, il processo di differenziazione sesso-specifica dei dotti genitali è preceduto dallo sviluppo, in entrambi gli embrioni XX e XY, di primordi sessualmente indifferenziati. Tali strutture, che originano dal mesonefro intorno alla VII settimana, sono rappresentate da due dotti pari e paralleli: il dotto mesonefrico di Wolff e il dotto para-mesonefrico di Müller.
Nel testicolo in via di sviluppo l’inizio della sintesi di AMH da parte delle cellule del Sertoli provoca la progressiva degenerazione e scomparsa dei dotti di Müller (17). Al contrario, il testosterone, che viene più tardi prodotto dalle cellule del Leydig, influenza positivamente lo sviluppo dei dotti di Wolff, che si differenziano in epididimi, vasi deferenti e vescichette seminali. A loro volta, gli epididimi si connettono al testicolo tramite i duttuli efferenti.
Durante lo sviluppo femminile non viene invece prodotto testosterone. Questo e probabilmente anche altri fattori provocano la regressione dei dotti di Wolff. Inoltre, in assenza di cellule del Sertoli produttrici di AMH, permangono i dotti di Müller. Dal loro successivo sviluppo, in parte mediato da WNT4 (18), originano le tube (porzione prossimale) e, distalmente, l'abbozzo utero-vaginale, da cui si formano l'utero e il terzo prossimale della vagina.
Sviluppo dei genitali esterni
I genitali esterni, anch’essi inizialmente identici in tutti i feti, indipendentemente da quale sia il sesso cromosomico o gonadico, si sviluppano dal tubercolo genitale, dalle pieghe uro-genitali e dai rigonfiamenti genitali (labio-scrotali). La formazione di queste strutture indifferenziate, che avviene tra l’VIII e la XII settimana gestazionale, è in parte mediata dal gene SHH e coinvolge la cascata WNT/β-catenina (19).
Nel feto di sesso maschile, sin dalla X-XIV settimana gestazionale e fino alla nascita, in presenza di adeguate quantità di diidro-testosterone (DHT), prodotto dalla conversione del testosterone ad opera dell’enzima 5 alfa-reduttasi, si viene a formare la prostata e si compie la progressiva mascolinizzazione dei genitali esterni (20): il tubercolo genitale dà origine al pene, le pieghe uro-genitali si fondono per formare l’uretra tubulare peniena e i rigonfiamenti genitali si uniscono medialmente per formare la sacca scrotale. In questa, durante l’ultimo trimestre di gestazione si posiziona il testicolo, dopo la sua discesa, in parte guidata da INLS3 prodotto dalle cellule del Leydig (21). Oltre a essere il maggior determinante della differenziazione dei genitali esterni maschili (22), DHT è il principale responsabile, dalla pubertà in poi, dello sviluppo dei caratteri secondari nel maschio normale (23,24). Appare comunque ormai evidente che la differenziazione dei genitali esterni maschili non rappresenta un processo unicamente androgeno-dipendente, ma è controllata anche da altri ormoni, in particolare dagli estrogeni, e vede anche coinvolti meccanismi ormono-indipendenti (25).
Nella femmina, in assenza dell’effetto degli androgeni, il tubercolo genitale si trasforma nel clitoride, le pieghe uro-genitali si sviluppano nelle piccole labbra e i rigonfiamenti genitali danno origine alle grandi labbra. Il meato uretrale si colloca nel perineo e, infine, si formano i due terzi distali della vagina. Tutti questi eventi sono influenzati dall’ambiente materno, abbondantemente estrogenizzato. Sebbene gli esatti meccanismi molecolari alla base della formazione dei genitali esterni non siano conosciuti in dettaglio, è indubbio che nell’acquisizione del fenotipo sessuale entrino in causa molti fattori e, tra questi, l’attività di diversi recettori steroidei (degli androgeni e degli estrogeni) e anche complesse interazioni epitelio-mesenchima, fondamentali per una coordinata e appropriata differenziazione dei genitali esterni (19).
BIBLIOGRAFIA
- Brennan J, Capel B. One tissue, two fates: molecular genetic events that underlie testis versus ovary development. Nat Rev Genet 2004, 5: 509-21.
- Wilhelm D, Palmer S, Koopman P. Sex determination and gonadal development in mammals. Physiol Rev 2007, 87: 1-28.
- MacLaughlin DT, Donahoe PK. Sex determination and differentiation. N Engl J Med 2004, 350: 367-78.
- Hughes IA. Intersex. BJU Int 2002, 90: 769-76.
- Neri G, Opitz J. Syndromal (and nonsyndromal) forms of male pseudohermaphroditism. Am J Med Genet 1999, 89: 201-9.
- Tanaka SS, Nishinakamura R. Regulation of male sex determination: genital ridge formation and Sry activation in mice. Cell Mol Life Sci 2014, 71: 4781-802.
- Eggers S, Ohnesorg T, Sinclair A. Genetic regulation of mammalian gonad development. Nat Rev Endocrinol 2014, 10: 673-83.
- Sinclair AH, Berta P, Palmer MS, et al. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 19: 240-4.
- Gubbay J, Collignon J, Koopman P, et al. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 1990, 346: 245-50.
- Stukenborg JB, Colón E, Söder O. Ontogenesis of testis development and function in humans. Sex Dev 2010, 4: 199-212.
- Schlessinger D, Garcia-Ortiz JE, Forabosco A, et al. Determination and stability of gonadal sex. J Androl 2010, 31: 16-25.
- Hughes IA. Female development--all by default? N Engl J Med 2004, 351: 748-50.
- Tevosian SG. Genetic control of ovarian development. Sex Dev 2013, 7: 33-45.
- Lin Y, Gill ME, Koubova J, Page DC. Germ cell-intrinsic and -extrinsic factors govern meiotic initiation in mouse embryos. Science 2008, 322: 1685-7.
- Chassot AA, Gregoire EP, Magliano M, et al. Genetics of ovarian differentiation: Rspo1, a major player. Sex Dev 2008, 2: 219-27.
- Eggers S, Sinclair A. Mammalian sex determination—insights from humans and mice. Chromosome Res 2012, 20: 215-38.
- Arango NA, Lovell-Badge R, Behringer RR. Targeted mutagenesis of the endogenous mouse Mis gene promoter: in vivo definition of genetic pathways of vertebrate sexual development. Cell 1999, 99: 409-19.
- Biason-Lauber A, Konrad D. WNT4 and sex development. Sex Dev 2008, 2: 210-8.
- Blaschko SD, Cunha GR, Baskin LS. Molecular mechanisms of external genitalia development. Differentiation 2012, 84: 261-8.
- Wilson JD, Griffin JE, George FW, Leshin M. The role of gonadal steroids in sexual differentiation. Recent Prog Horm Res 1981, 37: 1-39.
- Hughes IA, Acerini CL. Factors controlling testis descent. Eur J Endocrinol 2008, 159 suppl 1: S75-82.
- Wilson JD, George FW, Renfree MB. The endocrine role in mammalian sexual differentiation. Recent Prog Horm Res 1995, 50: 349-64.
- Rey RA, Grinspon RP. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract Res Clin Endocrinol Metab 2011, 25: 221-38.
- Bertelloni S, Dati E, Ghirri P, et al. Gestione clinica dei disturbi della differenziazione sessuale con cariotipo 46,XY: aspetti emergenti. Prospettive in Pediatria 2013, 41: 110-20.
- Weiss DA, Rodriguez E Jr, Cunha T, et al. Morphology of the external genitalia of the adult male and female mice as an endpoint of sex differentiation. Mol Cell Endocrinol 2012, 354: 94-102.