Cause endocrine di sleep apnea

Ernesto De Menis, Elena Callegari
UO Medicina Interna, Dipartimento Medicina Clinica, Ospedale Generale, Montebelluna
INTRODUZIONE
La sindrome della sleep apnea (SAS) rappresenta una condizione clinica che negli ultimi anni ha assunto notevole rilevanza per l’aumento di prevalenza, principalmente correlata all’aumento dell’obesità, per la stretta associazione con gli effetti legati alla sonnolenza diurna (incidenti, qualità di vita) e per le conseguenze negative cardiovascolari ed endocrino-metaboliche. La SAS determina alterazioni secretorie di diversi ormoni e conseguenti disturbi endocrino-metabolici, inoltre le terapie ormonali possono determinare o esacerbare la SAS anche per gli effetti diretti centrali degli ormoni (1,2).
Escludendo le malattie endocrino-metaboliche generali, come obesità, diabete mellito, PCOS, non è nota la percentuale di SAS dovuta a cause endocrine specifiche, quali ipotiroidismo e acromegalia. Certamente i pazienti affetti da SAS devono essere valutati clinicamente per la possibile presenza di queste malattie e, in caso di sospetto, adeguatamente indagati e trattati.
ACROMEGALIA
L’acromegalia è la causa endocrina specifica più strettamente associata alla SAS ed è stata anche la più indagata. La prevalenza di SAS risulta variabile in relazione alle diverse metodiche utilizzate per la diagnosi, ai diversi criteri di inclusione (acromegalia attiva e non attiva), ma globalmente circa il 70% dei pazienti presenta SAS (2). Una piccola proporzione di acromegalici presenta una SAS di tipo centrale, legata ad alterazioni del tono somatostatinergico o alla concomitante presenza di scompenso cardiaco. La grande maggioranza dei pazienti presenta invece una forma ostruttiva (OSAS) o mista.
La patogenesi della forma ostruttiva risulta legata alle note alterazioni morfologiche del paziente acromegalico, con modifiche anatomiche delle strutture ossee interessate, ipertrofia dei tessuti molli orofaringei, come dimostrato da studi cefalometrici, endoscopici, TC e RM; tuttavia, intervengono anche altri fattori, quali le alterazioni dei muscoli faringei e respiratori in generale (3). In analogia a quanto osservato in generale nella SAS, anche nell’acromegalico il rischio risulta correlato all’età e al BMI. Nella maggioranza degli studi la durata della malattia acromegalica rappresenta un fattore predisponente alla SAS. La correlazione fra livelli di GH e IGF-I e presenza di SAS risulta più incerta: sicuramente però questi parametri ormonali di attività di malattia risultano condizionare una maggiore gravità degli indici di SAS.
La presenza di SAS potrebbe aggravare altre comorbilità tipiche dell’acromegalico, in particolare la cardiopatia, le aritmie, l’ipertensione arteriosa e il dismetabolismo glucidico. Nell’acromegalico, in accordo a Consensus Internazionali (4), la SAS rappresenta una comorbilità che deve essere obbligatoriamente indagata, considerando la sua elevata prevalenza. Alla diagnosi vengono consigliati un’accurata indagine anamnestica, l’utilizzo di scale di valutazione della sonnolenza diurna (come EPSS) e soprattutto la polisonnografia (PSG). I tempi di attesa per l’esecuzione della PSG sembrano essere un limite nella gestione dell’acromegalico, per cui è stato anche suggerito un approccio operativo nella tempistica di esecuzione dell’esame (5).
Le differenti terapie dell’acromegalia risultano migliorare vari aspetti dell’OSAS, ad esempio riduzione dei tessuti molli orofaringei e miglioramento degli indici di gravità dell’OSAS (6). Tuttavia, in molti pazienti, specie affetti da forme gravi, non si osserva remissione dell’OSAS (2). Pertanto, nel follow-up è necessario eseguire un controllo della SAS sia con questionari che con PSG. Sebbene negli acromegalici, proprio per la presenza di anomalie anatomiche, si possa ipotizzare l’utilizzo di provvedimenti chirurgici specifici per il trattamento delle OSAS, ciò non è stato estesamente indagato. La terapia di scelta rimane pertanto la CPAP, che ha dimostrato di indurre miglioramenti anche sulle comorbilità, ad esempio migliorando la sensibilità insulinica (7).
IPOPITUITARISMO
Non esistono dati riguardo all’associazione di ipopituitarismo in generale e SAS. Due condizioni cliniche risultano specificamente associate a SAS, la sindrome di Prader-Willi (PWS) e il craniofaringioma, in entrambe le quali l’obesità ha un ruolo molto rilevante.
Sindrome di Prader-Willi (PWS)
I pazienti con PWS hanno un’elevata frequenza di SAS: può essere presente una forma centrale, ma domina soprattutto la forma ostruttiva, legata principalmente all’obesità, ma anche all’ipotonia dei muscoli faringei. Nel 40-100% dei pazienti è presente deficit di GH, non legato alla concomitante presenza di obesità (8), con autorizzazione alla terapia sostitutiva con rhGH. Questa ha dimostrato effetti positivi nei pazienti con PWS (8), tuttavia, subito dopo l’introduzione della terapia con rhGH, erano stati segnalati casi di morte legati a problemi respiratori e di morti improvvise, facendo ipotizzare una correlazione fra rhGH e aggravamento della SAS. Il meccanismo potrebbe essere lo sviluppo di ipertrofia del tessuto linfatico adenoideo/tonsillare e la ritenzione idrosalina, con ispessimento dei tessuti molli peri-faringei; il GH potrebbe però migliorare l’ipoventilazione di origine centrale (9). Pertanto, prima di intraprendere la terapia con rhGH, le linee guida internazionali (10) consigliano la valutazione specialistica ORL per la presenza di ostruzione faringea (tonsille, adenoidi) e l’eventuale intervento, nonchè l’esecuzione di una PSG per valutare la presenza di OSAS. La terapia con rhGH è controindicata in presenza di OSAS severa non trattata. La terapia sostituiva con rhGH si accompagna in generale a un miglioramento dei parametri PSG, ma alcuni pazienti possono presentare un peggioramento (9), per cui viene consigliato di ripetere la PSG dopo 3-6 mesi e la terapia deve essere sospesa se compare OSAS dopo l’inizio di terapia e in caso di infezioni respiratorie alte (10).
Craniofaringioma
Fra le cause di morte dei pazienti con craniofaringioma sono riportate le patologie polmonari, inclusa l’OSAS. In questi pazienti sono frequentemente presenti disturbi del sonno, legati ad alterazioni del ritmo sonno-veglia, che comportano sonnolenza diurna. Inoltre, OSAS è una condizione frequente attribuita alla presenza di obesità. Da segnalare che le OSAS nei pazienti con craniofaringioma risultano di maggiore severità rispetto ai pazienti con uguale grado di obesità non dovuta a craniofaringioma (11). Pertanto, nei soggetti con craniofaringioma deve essere eseguita una valutazione clinica della sonnolenza diurna, con l’esecuzione di PSG nei soggetti con sospetto o a rischio di OSAS.
IPOTIROIDISMO
L’ipotiroidismo e l’OSAS sono condizioni relativamente frequenti nella popolazione generale e, sebbene l’associazione di queste due condizioni sia nota da molto tempo, i dati epidemiologici sono limitati. Studi retrospettivi e prospettici nei pazienti ipotiroidei hanno dimostrato una prevalenza di SAS variabile da 25% a oltre 50%. Viceversa la prevalenza di ipotiroidismo nei pazienti valutati per OSAS varia da 1 a 3% (1,12,13). Questo ha indotto a consigliare lo screening della funzione tiroidea in tutti i pazienti con OSAS, mentre per altri ciò non è giustificato in assenza di quadro clinico suggestivo. Sono stati descritti casi di SAS centrale, legata all’effetto degli ormoni tiroidei sulla risposta ventilatoria centrale, ma la maggior parte dei casi sono OSAS.
Il meccanismo chiamato in causa è un’alterazione dei tessuti molli faringei, con accumulo di mucopolisaccaridi; potrebbe intervenire anche la miopatia da ipotiroidismo. I pazienti ipotiroidei con OSA hanno età maggiore di quelli senza e sicuramente hanno maggior BMI. Rari casi di OSAS sono legati alla presenza di voluminoso gozzo.
Il trattamento dell’ipotiroidismo determina sicuramente un netto miglioramento degli indici di OSAS e della sintomatologia clinica, per cui prima di iniziare la terapia con CPAP nei pazienti con ipotiroidismo è opportuno attendere gli effetti della terapia sostitutiva.
Le conclusioni sono che ogni paziente ipotiroideo deve essere indagato sulla possibile sintomatologia connessa a OSAS, mentre non è indicato screening per ipotiroidismo nei pazienti con OSAS senza quadro clinico di sospetto.
BIBLIOGRAFIA
- Saaresranta T, Polo D. Sleep-disordered breathing and hormones. Eur Resp J 2003, 22: 161–72.
- Attal P, Chanson P. Endocrine aspects of obstructive sleep apnea. J Clin Endocrinol Metab 2010, 95: 483–95.
- Castellani C, et al. Morphological study of upper airways and long-term follow-up of obstructive sleep apnea syndrome in acromegalic patients. Endocrine 2016, 51: 308-16.
- Melmed S, et al. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary 2013, 16: 294-302.
- De Menis E, et al. Assessment of the awareness and management of sleep apnea syndrome in acromegaly. The COM.E.TA (Comorbidities evaluation and treatment) Italian study group. J Endocrinol Invest 2011, 34: 60-4.
- Berg C, et al. Influence of disease control with pegvisomant on sleep apnoea and tongue volume in patients with active acromegaly. Eur J Endocrin 2009, 161: 829-35.
- Duarte FH, et al. The impact of sleep apnea treatment on carbohydrate metabolism in patients with acromegaly. Pituitary 2013, 16: 341-50.
- Aycan Z, Bas VN. Prader-Willi Syndrome and Growth Hormone Deficiency. J Clin Res Pediatr Endocrinol 2014, 6: 62-7.
- Miller J, et al. Short-term effects of growth hormone on sleep abnormalities in Prader-Willi syndrome. J Clin Endocrinol Metab 2006, 91: 413–7.
- Deal CL, et al. Growth Hormone Research Society Workshop Summary: Consensus Guidelines for Recombinant Human Growth Hormone Therapy in Prader-Willi Syndrome. J Clin Endocrinol Metab 2013, 98: E1072–87.
- O’Gorman CS, et al. Sleep-disordered breathing is increased in obese adolescents with craniopharyngioma compared with obese controls. J Clin Endocrinol Metab 2010, 95: 2211–8.
- Rajagopal KR, et al. Obstructive sleep apnea in hypothyroidism. Ann Intern Med 1984, 101: 491-4.
- Lin CC, et al. The relationship between sleep apnea syndrome and hypothyroidism. Chest 1992, 102: 1663-7.
Manifestazioni endocrino-metaboliche di sleep-apnea
Cinzia Castellani & Maria Vittoria Davì
UOS di Endocrinologia, Medicina generale e malattie aterotrombotiche e degenerative, Policlinico GB Rossi, AOUI Verona
La sindrome delle apnee notturne è la forma più frequente tra i disturbi respiratori nel sonno, definita da un insieme di segni e sintomi, tra cui spiccano russamento intermittente, sonnolenza diurna e riduzione delle performance diurne.
Sul piano fisiopatologico è caratterizzata da ripetuti e frequenti episodi di apnea o ipopnea (minimo 5 episodi/ora), responsabili di ipossiemia con desaturazioni arteriose di ossigeno di diversa entità, seguiti da risvegli notturni più o meno completi. A seconda dell'eziologia, meccanica o nervosa, le apnee si distinguono in ostruttive o centrali, ma essendo le prime più frequenti, si può parlare semplicemente di OSAS, acronimo inglese di Obstructive Sleep Apnea Syndrome.
La rilevanza clinica di tale disturbo è diffusamente riconosciuta, in quanto associato a un aumentato rischio di incidenti stradali dovuti a sonnolenza diurna per sonno frammentato, a una maggior predisposizione allo sviluppo di patologie cardio-vascolari, oltre che ad alterazioni metaboliche e ormonali. Queste ultime consistono principalmente in aumento dell'insulino-resistenza, dell'attività simpatico-adrenergica, della cortisolemia e nella disfunzione sessuale.
È noto che l'obesità, principalmente viscerale, è un fattore di rischio per lo sviluppo delle apnee notturne, e, allo stesso tempo, i pazienti affetti da OSAS tendono ad aumentare di peso o presentano difficoltà nel calo ponderale. Le cause sembrano essere lo sviluppo di iperattività simpatica e l'aumento dei livelli di leptina, a cui si associa il fenomeno della cosiddetta leptino-resistenza, ossia un inadeguato segnale di riduzione del grasso corporeo nonostante l'aumento dell'ormone in questione. La leptino-resistenza, tuttavia, si manifesta solo da un punto di vista metabolico, mentre vengono mantenuti gli effetti stimolanti della leptina sul sistema cardio-vascolare e nervoso simpatico (1).
L'OSAS, inoltre, rappresenta di per sé una condizione di stress per l'organismo, soprattutto a causa dei continui risvegli notturni associati: viene quindi favorita un'iperattivazione dell'asse ipotalamo-ipofisi-surrene, con conseguente sviluppo di ipercortisolemia. Tale meccanismo sembra essere alla base dell'aumento della leptina, dello sviluppo di ipertensione arteriosa, causata anche da un aumentato rilascio di aldosterone, dell'insorgenza di intolleranza glucidica, legata altresì alla deprivazione di sonno, oltre che allo sviluppo di insulino-resistenza e dell'accumulo di grasso viscerale addominale, a sua volta predisponente tale alterazione metabolica (2,3). L'insulino-resistenza nella sindrome delle apnee notturne sembra essere favorita anche dalla presenza di ipossiemia intermittente e di iperattività simpatica, scatenata dalla deprivazione di sonno (4). Si può quindi affermare che l'OSAS è un fattore di rischio indipendente per lo sviluppo di diabete mellito tipo 2.
La sonnolenza diurna, conseguente alle apnee notturne, determina inoltre un aumentato rilascio delle citochine pro-infiammatorie, in particolare di tumor necrosis factor (TNF)-α e interleukina (IL)-6, coinvolte sia nella regolazione fisiologica del sonno, sia nella patogenesi dell'infiammazione sistemica nell'OSAS (5), con un'aumentata produzione di radicali liberi dell'ossigeno. Anche la proteina C-reattiva ultrasensibile è elevata nei pazienti con tali disturbi del sonno: oltre ad essere associata al tessuto adiposo viscerale, essa è significativamente correlata con la presenza di insulino-resistenza (6). Si può pertanto spiegare lo sviluppo e la correlazione tra infiammazione sistemica, insulino-resistenza, obesità e aterosclerosi, alla base delle implicazioni metaboliche nei pazienti con OSAS.
Nei soggetti affetti da sindrome delle apnee notturne, l'insulino-resistenza, insieme all'ipossia, sembrano giustificare anche lo sviluppo del danno epatico, con conseguente incremento degli enzimi epatici, che si estrinseca con alterazioni che spaziano dallo sviluppo di steatosi, alla necrosi lobulare, fino alla cirrosi. La prevalenza della steato-epatite non alcolica (NASH) sembra essere correlata con il grado di severità dell'OSAS, valutabile con l'indice apnea-ipopnea (AHI); tuttavia sono necessari ulteriori studi a riguardo, soprattutto per valutare eventuali benefici del trattamento dell'OSAS sul danno epatico insorto (7).
Per quanto riguarda le alterazioni dell'asse ipotalamo-ipofisi, nei pazienti con sindrome delle apnee notturne, a causa dell'ipossia e dell'alterata regolazione nervosa, si sviluppa un'aumentata funzione corticotropa, associata a una riduzione di quella somatotropa. In questi pazienti, infatti, si può documentare un'aumentata risposta dell'ACTH al CRH e una ridotta secrezione e sensibilità del GH: rispetto ai pazienti semplicemente obesi, in caso di OSAS si osserva, infatti, una più marcata compromissione della capacità secretoria massima delle cellule somatotrope e una ridotta sensibilità dell'IGF-1 alla stimolazione da parte del GH. Nei soggetti affetti da sindrome delle apnee notturne non sembrano invece rilevarsi alterazioni nella risposta di TSH e PRL alla stimolazione del TRH (8).
In pazienti maschi affetti da OSAS, di mezza età, si può documentare, inoltre, uno stato di ipoandrogenismo: la compromissione dell'asse gonadico, con riduzione della secrezione di LH e di quella pulsatile del testosterone, è maggiore in questi soggetti rispetto ai semplici obesi a causa dell'ipossia, degli aumentati livelli di leptina e della frammentazione del sonno, indipendentemente dall'età, dal BMI e dal grasso viscerale (9). La terapia con CPAP può aumentare i livelli di testosterone, rendendo reversibile la disfunzione ipotalamo-ipofisi-gonadi dovuta alla deprivazione del sonno (10). Secondo le linee guida dell’Endocrine Society del 2010 sul trattamento con testosterone nell’ipogonadismo, la terapia sostitutiva con testosterone (TRT) nei pazienti con OSAS grave non trattata è controindicata (11). Infatti, la TRT sembra possa alterare la risposta ventilatoria mediante l’effetto sui chemorecettori centrali. Tuttavia, recenti studi dimostrano che, se associata al trattamento con CPAP, la TRT può non solo migliorare l'ipogonadismo, ma alleviare la disfunzione erettile/sessuale (12). Di conseguenza, la recente rivisitazione delle linee guida del 2010 ha rifiutato il dogma contro la terapia con TRT nell’OSAS non trattato (13).
Anche nelle donne, l'OSAS è associata ad una significativa diminuzione della funzione sessuale, valutata da un punto di vista fisico, psichico e ormonale; tuttavia, la gravità delle apnee notturne non sembra correlare con il grado della disfunzione sessuale femminile (FSD). Per questa ragione, verosimilmente altre comorbilità, sia organiche, come un aumentato BMI, che psicofisiche, come sonnolenza, stanchezza e depressione, sembrano essere i fattori che contribuiscono allo sviluppo di FSD nelle donne con OSAS (14).
L'OSAS, quindi, oltre a essere una conseguenza della sindrome metabolica e di varie alterazioni ormonali, ne può essere a sua volta causa; di conseguenza, è importante ricercare questo disturbo del sonno nei casi sospetti, per intraprendere, se necessario, una terapia specifica, al fine di evitare o ridurre le importanti complicanze di tipo cardio-vascolare, metabolico e ormonale.
Bibliografia
- Phillips BG, Kato M, Narkiewicz K, et al. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am J Physiol Heart Circ Physiol 2000, 279: H234-7.
- Buckley TM, Schatzberg AF. On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: normal HPA axis activity and circadian rhythm, exemplary sleep disorders. J Clin Endocrinol Metab 2005, 90: 3106-14.
- Ip MS, Lam B, Ng MM, et al. Obstructive sleep apnea is independently associated with insulin resistance. Am J Respir Crit Care Med 2002, 165: 670-6.
- Tasali E, Van Cauter E. Sleep-disordered breathing and the current epidemic of obesity. Am J Respir Crit Care Med 2002, 165: 562–3.
- Vgontzas AN, Bixler EO, Chrousos GP. Metabolic disturbances in obesity versus sleep apnoea: the importance of visceral obesity and insulin resistance. J Intern Med 2003, 254: 32-44.
- Shinji T, Hiroshi Y, Yasuhiro Y, et al. Obstructive sleep apnea causes systemic inflammation and metabolic syndrome. Chest 2005, 127: 1074-5.
- Tannè F, Gagnadoux F, Chazouilleres O, et al. Chronic liver injury during obstructive sleep apnea. Hepatology 2005, 41: 1290-6.
- Lanfranco F, Gianotti L, Pivetti S, et al. Obese patients with obstructive sleep apnoea syndrome show a peculiar alteration of the corticotroph but not of the thyrotroph and lactotroph function. Clin Endocrinol 2004, 60: 41-8.
- Luboshitzky R, Lavie L, Shen-Orr Z, et al. Altered luteinizing hormone and testosterone secretion in middle-aged obese men with obstructive sleep apnea. Obes Res 2005, 13: 780-6.
- Grunstein RR, Handelsman DJ, Lawrence SJ, et al. Neuroendocrine dysfunction in sleep apnea: reversal by continuous positive airways pressure therapy. J Clin Endocrinol Metab 1989, 68: 352-8.
- Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010, 95: 2536-59.
- Burschtin O, Wang J. Testosterone deficiency and sleep apnea. Urol Clin North Am 2016, 43: 233-7.
- Seftel AD, Kathrins M, Niederberger C. Critical update of the 2010 Endocrine Society clinical practice guidelines for male hypogonadism: a systematic analysis. Mayo Clin Proc 2015, 90: 1104-15.
- Onem K, Erol B, Sanli O, et al. Is sexual dysfunction in women with obstructive sleep apnea-hypopnea syndrome associated with the severity of the disease? A pilot study. J Sex Med 2008, 5: 2600-9.
Ripercussioni endocrine dell’infezione da HIV e delle terapie anti-retrovirali
Paolo Zuppi
Endocrinologia San Camillo-Forlanini, Roma
INTRODUZIONE (1)
L’HIV (Human Immunodeficiency Virus) è un retrovirus, che infetta le cellule che esprimono il recettore CD4, proteina di membrana presente sui linfociti T-Helper. Una volta penetrato nel citoplasma cellulare, grazie alla trascrittasi inversa, una DNA-polimerasi RNA-dipendente, sintetizza DNA dal RNA virale e lo integra nel DNA della cellula.
L’infezione virale direttamente e indirettamente distrugge i linfociti T-helper, provocando una sindrome da deficienza del sistema immunitario. AIDS (Acquired Immune Deficiency Syndrome) è una malattia cronica sistemica multiorgano, caratterizzata da infezioni opportunistiche e patologie neoplastiche.
La trasmissione del virus avviene per via sessuale, parenterale e verticale (da madre a figlio durante la gravidanza). Se non curata, l’infezione determina la morte in circa 10 anni. Il quadro sindromico è in continuo mutamento per le incessanti innovazioni farmacologiche ed estremamente differente in base alla possibilità di accesso ai farmaci.
Epidemiologia (2)
La malattia da HIV è una pandemia: sono valutati circa 33.000.000 di infetti nel mondo. Negli ultimi anni, grazie alle campagne di informazione e di prevenzione e alla maggiore disponibilità dei farmaci, sono diminuiti sia il numero di nuovi infetti che di morti per AIDS. Si ritiene che in Italia vi siano circa 125.000 persone HIV+, con una prevalenza intorno allo 0.28% e un’incidenza di 6 nuovi casi/anno per 100.000 residenti.
L’età media della popolazione affetta è in continuo aumento, non solo per l’efficacia dei nuovi farmaci ma anche per la più elevata età al contagio.
Terapia (3)
L’HAART (Highly Active Antiretroviral Therapy) consiste nell'assunzione combinata di farmaci anti-retrovirali. In base al meccanismo di azione, i farmaci sono classificati in:
- inibitori non-nucleosidici della trascrittasi inversa (NNRTI);
- inibitori nucleosidici della trascrittasi inversa (NRTI);
- inibitori della proteasi (PI);
- inibitori della fusione;
- antagonisti CCR5;
- inibitori dell'integrasi.
L’HAART ha determinato un sostanziale miglioramento della durata e della qualità della vita dei pazienti, modificando profondamente la malattia da HIV. La letteratura sugli effetti collaterali e sulle interazioni dei numerosi farmaci assunti da questi pazienti deve essere aggiornata continuamente per l’incessante segnalazione di nuove evidenze ed è quindi opportuno consultare il Drug Interaction Checker dell’Università di Liverpool (4). La farmacogenetica può aiutare la personalizzazione della terapia e quindi a ridurre gli effetti indesiderati.
RIPERCUSSIONI ENDOCRINE
Saranno qui trattati essenzialmente gli aspetti clinici riscontrabili nei pazienti HIV con possibilità di accesso alle cure.
Le manifestazioni cliniche dall’infezione da HIV sono riconducibili non solo all’effetto diretto del virus, alle infezioni e neoplasie concomitanti da immunodeficienza, alla terapia HAART, per la sua efficacia (immune reconstitution inflammatory syndrome, IRIS), per gli effetti collaterali e per le interazioni con altri farmaci, ma anche all’uso di sostanze a scopo voluttuario, alla malnutrizione, alle cattive abitudini di vita e allo stress psicologico. Il paziente HIV+ deve essere valutato nella sua complessità.
Ipofisi (5)
Negli studi dei primi anni '90, i pazienti HIV+ presentavano all’autopsia processi infettivi e/o neoplastici ipofisari in circa il 10% dei casi. Attualmente l’incidenza è ridotta ed è rara l’evidenza di ipopituitarismo.
È stata proposta la terapia con GH e più recentemente con tesamorelina, alla luce dei suoi possibili vantaggi su wasting syndrome, lipodistrofia, immunodeficienza e osteoporosi. In Italia, rhGH è prescrivibile a carico del SSN solo nei pazienti con dimostrato deficit.
Iposodiemia (6)
Iponatremia è presente in circa il 50% dei pazienti con AIDS ricoverati e nel 20% dei pazienti domiciliari. I fattori eziopatogenetici possono essere perdite gastrointestinali, ipofunzione surrenalica e resistenza ai glucocorticoidi, danno renale, SIAD (syndrome of inappropriate antidiuretic hormone secretion) e CWS (cerebral salt wasting syndrome), farmaci.
La delicata gestione dell’iposodiemia è basata sugli stessi principi dei pazienti non HIV.
Tiroide (7)
I pazienti HIV+ presentano una riduzione di FT3 correlata con la gravità della malattia. La prevalenza di patologie tiroidee infiammatorie è aumentata rispetto alla popolazione normale, in particolare nei pazienti che, assumendo HAART, hanno una riacquisita competenza immunitaria. Non appare comunque indicato uno screening della funzionalità tiroidea nei pazienti asintomatici. Nei pazienti tubercolari la rifampicina può slatentizzare un ipotiroidismo preclinico o richiedere l’incremento posologico della L-T4 nei pazienti già in terapia sostitutiva.
Surreni (8)
La complessa interazione fra asse ipotalamo-ipofisi-surrene e sistema immunitario è profondamente alterata nei pazienti affetti da malattia da HIV. Conosciamo solo minimamente i meccanismi che alterano questo crocevia di diversi sistemi: ad esempio, una proteina virale, determinando l’aumento della trascrizione del recettore per i glicocorticoidi, può provocare ipersensibilità ai glicocorticoidi.
La cortisolemia è dapprima normale poi, spesso, aumentata; i livelli degli androgeni surrenalici sono ridotti. Vi è una progressiva riduzione dei livelli di DHEAS, che correlano significativamente con i livelli dei CD4 e che sembrano predittivi della progressione della malattia.
La terapia con DHEA per os sembra migliorare la depressione e il senso di benessere.
Gli inibitori della proteasi, in particolare il ritonavir, inibendo la CYP3A4, enzima necessario all’inattivazione di farmaci steroidei, determinano sindrome di Cushing in seguito a uso di steroidi anche locale e insufficienza surrenalica secondaria alla sospensione.
Farmaci frequentemente utilizzati in questi pazienti possono interferire: ad esempio il chetoconazolo inibisce la steroidogenesi, rifampicina e fenitoina accelerano la degradazione del cortisolo.
Lo screening per l’insufficienza surrenalica non è consigliato di routine, ma solo nei pazienti che presentino anoressia, nausea, calo ponderale, astenia, ipotensione, iponatremia, infezione da citomegalovirus o tubercolare, e nei pazienti che debbano essere sottoposti a interventi chirurgici o siano ricoverati in terapia intensiva.
Gonadi maschili (9)
L’HAART ha ridotto l’incidenza dell’ipogonadismo dal 50% al 20%; tuttora però circa il 60% dei pazienti HIV+ lamentano riduzione della libido e della potenza sessuale. Tale situazione è legata alle comorbilità, in particolare cachessia, malattie sistemiche, cattive abitudini di vita, sostanze voluttuarie, stress emotivo e psicologico, disordini neurologici, effetti collaterali o interazione di farmaci.
I pazienti HIV+ presentano un aumento della capacità legante il testosterone, per cui è consigliato il calcolo della quota libera del testosterone. Fra i pazienti HIV+, molti assumono terapia con androgeni senza evidenza laboratoristica di ipogonadismo. Il sildenafil interagisce a livello del citocromo P450 3A4 con il saquinavir e il ritonavir, per cui è consigliato iniziare con basse dosi.
Gonadi femminili (10)
Estradiolo e progesterone alterano la trascrizione virale, per cui nelle donne in età fertile la carica virale risulta minore. Sono frequentemente presenti alterazioni del ciclo mestruale legate alle comorbilità e alle condizioni generali. L’età media della menopausa sembrerebbe essere ridotta, ma cicli anovulatori e amenorrea prolungata pongono un difficile problema di diagnosi differenziale.
Riproduzione (11,12)
Nelle coppie sierodiscordanti, la terapia HAART assunta dall’infetto determina una notevole riduzione del rischio di contagio per il partner sano, grazie alla riduzione della carica virale, essendo quasi a zero per cariche < 50 copie/mL; comunque, il trattamento preventivo del partner non infetto è altamente efficace.
Nelle coppie sierodiscordanti con maschio infetto che desiderino una gravidanza, la tecnica dello sperm washing è sicura ed efficace.
L’incidenza di eventi avversi in gravidanza è significativamente aumentata ed è in relazione alla carica virale e alle patologie concomitanti. Recentemente è stato segnalato un rischio di gestosi modestamente aumentato nelle donne in terapia HAART.
Osso (13)
I pazienti HIV+ hanno aumentata incidenza di osteoporosi e aumentato rischio di fratture. La perdita di massa ossea è proporzionale all’età, alla riduzione del BMI e alla viremia. I momenti eziofisiopatogenetici sono molteplici: proteine virali, citochine infiammatorie, comorbilità infettive (in particolare HCV), HAART, altri farmaci, inattività fisica, fumo di sigaretta, ipogonadismo, malnutrizione, calo ponderale/cachessia. Inoltre, nei pazienti HIV+ è spesso presente ipovitaminosi D.
L’HAART, all’inizio, sembra provocare un peggioramento della condizione ossea, per poi determinare un lento miglioramento in relazione a quello delle condizioni generali; infatti, la prevalenza dell’osteoporosi, aggiustata per altri fattori di rischio, resta comunque aumentata nei pazienti in HAART. I farmaci maggiormente dannosi sono il tenofovir, della classe NRTI e gli inibitori della proteasi; meno dannosi sembrerebbero gli inibitori dell’integrasi. Vi sono peraltro complesse interazioni ancora non conosciute, ad esempio zidovudina ed efavirenz determinano riduzione della vitamina D.
Lo screening con DEXA è consigliato nelle seguenti condizioni: storia di fratture a basso impatto, donne in post-menopausa, maschi > 50 anni, familiarità per frattura di femore, aumentato rischio di cadute, ipogonadismo, assunzione prolungata di steroidi, bassi CD4, HAART, HCV+, fumo di sigaretta, cachessia.
La base del trattamento è la correzione dei fattori predisponenti e la somministrazione di vitamina D. I farmaci con evidenze di efficacia, anche se limitate, sono alendronato per os settimanale e zoledronico ev annuale. Non vi sono ancora evidenze su denosumab e teriparatide. La rosuvastatina sembrerebbe modestamente efficace grazie alla riduzione della flogosi. L’AIFA prevede nella nota 79 la dispensazione a carico del SSN dei farmaci in prevenzione primaria per i pazienti affetti da AIDS nelle donne in menopausa o negli uomini di età ≥ 50 anni, con un T-score colonna o femore ≤ -3.
Metabolismo glicolipidico (14)
Nei pazienti HIV+ sono frequenti alterazioni del metabolismo glicolipidico e lipodistrofia. I meccanismi di queste alterazioni sono molto complessi e ancora poco conosciuti. In questi pazienti, oltre ai fattori eziopatogenetici noti (familiarità, tabagismo, vita sedentaria, obesità, ecc) hanno rilevanza l’infiammazione cronica, le disfunzioni immunitarie ma soprattutto l’HAART. La dislipidemia, presente in circa il 70% dei pazienti in terapia, è particolarmente associata all’assunzione di NRTI, NNRTI e PI. La lipodistrofia può essere caratterizzata da accumulo di grasso viscerale (lipoipertrofia), lipomatosi distrettuale (gobba di bufalo) o lipoatrofia del sottocutaneo, specie del volto e degli arti. L’assunzione di farmaci anti-retrovirali si associa a maggior rischio di lipodistrofia, in particolare la variante lipoatrofica si associa all’uso degli NRTI.
Nei pazienti affetti da malattia da HIV è necessario raccogliere i dati antropometrici, BMI, circonferenza vita e valutare accuratamente la topografia del tessuto adiposo. Lo screening delle patologie metaboliche prevede: identificazione della familiarità, valutazione dei fattori di rischio tradizionali, glicemia a digiuno, assetto lipidico, insulinemia. Ove il paziente venga posto in HAART, questi parametri di laboratorio devono essere rivalutati già dopo 4-8 settimane, altrimenti, in assenza di alterazioni, è indicato un controllo annuale.
La proposta terapeutica deve tenere in considerazione gli aspetti comportamentali e sociali di ogni paziente. La base della terapia è il cambiamento dello stile di vita: sospensione del fumo, incremento dell’attività fisica di tipo aerobico, dieta ipocalorica con riduzione dei grassi saturi e astensione dall’alcool. È dimostrato che questi provvedimenti provocano una riduzione del grasso viscerale e un miglioramento della sensibilità insulinica e delle frazioni lipidiche. Se le modificazioni dello stile di vita si dimostrano insufficienti, prima di un intervento farmacologico è opportuno rivalutare collegialmente la terapia anti-retrovirale. Nei pazienti HIV+, il farmaco di prima scelta per il diabete è la metformina, alla dose iniziale di 500-850 mg/die, con eventuale incremento fino a 2-3 g/die. La metformina può determinare un peggioramento della lipoatrofia. Il pioglitazone è indicato nel soggetto non obeso, poiché determina miglioramento della resistenza all’insulina, dell’assetto lipidico e della lipoatrofia. Per altri farmaci anti-diabetici non vi sono dati forti nella popolazione HIV. I fibrati sono indicati nell’ipertrigliceridemia > 500 mg/dL. Tra le statine, bisogna evitare lovastatina e sinvastatina per la loro interazione con i PI e preferire pravastatina e rosuvastatina, comunque utilizzando la dose minima terapeutica e monitorando il possibile danno epatico e muscolare. I farmaci NNRTI, in particolare l’efavirenz, accelerano il metabolismo delle statine.
Gli accumuli lipomatosi (gobba di bufalo) possono essere corretti chirurgicamente e la lipoatrofia del volto con autotrapianto di tessuto adiposo o con fillers riassorbibili o permanenti, sebbene, purtroppo, i risultati a distanza di tali interventi non siano sempre soddisfacenti.
CONCLUSIONI
La malattia da HIV è una malattia cronica sistemica con un quadro sindromico complesso risultato di molteplici fattori, la cui espressione clinica è in continuo divenire in relazione a nuovi farmaci e all’invecchiamento della popolazione. Abbiamo una conoscenza molto limitata dei meccanismi fisiopatologici. Le patologie d’organo sono profondamente influenzate dalle condizioni generali. Il paziente HIV+ deve essere seguito nella sua complessità con una terapia personalizzata da una equipe dedicata. La creazione di una buona relazione è la base indispensabile all’efficacia della terapia. È necessario contemplare il paziente nella sua totalità, considerando le sue caratteristiche esistenziali, i fattori socioculturali e il vissuto della malattia.
BIBLIOGRAFIA ESSENZIALE
- United Nations. AIDS. 2015 (consultato il 24-05-2016).
- Camoni L, Regine V, Stanecki K, et al. Estimates of the number of people living with HIV in Italy. Biomed Res Int 2014, 2014: 209619.
- Linee guida su utilizzo antiretrovirali in persone con infezione HIV-1. 2012. (consultato il 24-05-2016).
- University of Liverpool. Drug Interaction Checker: HIV drug interactions (consultato il 27-05-2016).
- University of California San Francisco. HIV insite (consultato il 23-05-2016).
- Menon MC, Garcha AS, Khanna A. The management of hyponatremia in HIV disease. J Nephrol 2014, 27: 109.
- Weetman AP. Thyroid abnormalities. Endocrinol Metab Clin North Am 2014, 43: 781-90.
- Kino T. AIDS/HPA Axis. 2012 Nov 12. In: De Groot LJ, Beck-Peccoz P, Chrousos G, et al, editors. Endotext.
- Rietschel P, Corcoran C, Stanley T, et al. Prevalence of hypogonadism among men with weight loss related to human immunodeficiency virus infection who were receiving highly active antiretroviral therapy. Clin Infect Dis 2000, 31: 1240-4.
- Yalamanchi S, Dobs A, Greenblatt RM. Gonadal function and reproductive health in women with human immunodeficiency virus infection. Endocrinol Metab Clin North Am 2014, 43: 731-41.
- Loutfy MR, Wu W, Letchumanan M, et al. Systematic review of HIV transmission between heterosexual serodiscordant couples where the HIV-positive partner is fully suppressed on antiretroviral therapy. PLoS One 2013, 8: e55747.
- Senise J, Bonafe S, Castelo A. The management of HIV-infected pregnant women. Curr Opin Obstetr Gynecol 2012, 24: 395–401.
- European AIDS Clinical Society. Guidelines version 8.0. October 2015 (consultato il 24-05-2016).
- Lake JE, Currier JS. Metabolic disease in HIV infection. Lancet Infect Dis 2013, 13: 964-75.
Ripercussioni endocrine dei trapianti d'organo
Matteo Parolin, Francesca Dassie, Marialberta Battocchio, Pietro Maffei
Clinica Medica 3^, Dipartimento di Medicina (DIMED), Azienda Ospedaliera Padova
Farmaci immuno-soppressivi
Il successo della trapiantologia è dovuto anche all’efficacia della terapia farmacologica immunosoppressiva. Questa categoria di farmaci comprende classicamente glucocorticoidi, inibitori della calcineurina (ciclosporina e tacrolimus) e azatioprina. Per ridurre l’uso e gli effetti collaterali di quest’ultimi, attualmente vengono impiegati anche nuovi immunosoppressori, quali il micofenolato mofetile e gli inibitori di mTOR (sirolimus ed everolimus). Molti di questi farmaci possono produrre alterazioni endocrino-metaboliche.
Come noto, i glucocorticoidi possono produrre insulino-resistenza, iperglicemia e diabete (9-24% dei casi), in funzione di dose, durata della terapia, età, familiarità, etnia. È stata inoltre segnalata dislipidemia, con grado variabile in relazione ai diversi organi trapiantati (rene, fegato, cuore). Altro effetto non trascurabile è dato dalla soppressione della funzione surrenalica. Ricordiamo infine altri potenziali effetti collaterali di questa terapia: irsutismo, aumento ponderale, disionie, ipocalcemia, ritardo staturale, osteopenia e osteoporosi, ipertensione.
Il trattamento con ciclosporina può comportare iperlipidemia (frequentemente), anoressia, iperuricemia, iperpotassiemia, ipomagnesiemia e iperglicemia (raramente); raramente è stata descritta ginecomastia. Nel lungo termine il danno renale, l’ipovitaminosi D e l’iperparatiroidismo secondario favoriscono indirettamente la perdita di massa ossea. La terapia con tacrolimus si può invece associare frequentemente a iperglicemia, diabete e iperpotassiemia; raramente è stato descritto irsutismo; fra gli altri effetti indesiderati ricordiamo anche ipomagnesiemia, ipofosfatemia, ipocalcemia, iposodiemia, iperuricemia, anoressia, acidosi metabolica, ipercolesterolemia e ipertrigliceridemia; frequentemente viene descritta ipertensione. Anche con questo farmaco il danno renale può indurre secondariamente osteoporosi.
Il trattamento con micofenolato mofetile può determinare acidosi, iper e ipopotassiemia, iperglicemia, ipomagnesiemia, ipocalcemia, ipercolesterolemia, ipofosfatemia, iperuricemia e gotta, anoressia, ipertensione.
Durante il trattamento con sirolimus è stata descritta la comparsa di ipopotassiemia, ipofosfatemia, ipercolesterolemia e ipertrigliceridemia, iperglicemia e diabete, ipertensione, cisti ovariche, amenorree e menorragia. È stato inoltre descritto che gli inibitori di mTOR determinano una riduzione del testosterone con elevazione di LH, riduzione della spermatogenesi ed elevazione di FSH. Segnaliamo infine che la terapia con everolimus si può frequentemente associare ad anoressia, iperglicemia (meno frequentemente diabete mellito) e ipercolesterolemia (meno frequentemente ipertrigliceridemia); sono state inoltre descritte ipofosfatemia, ipopotassiemia, ipocalcemia, ipertensione, irregolarità del ciclo mestruale.
Trapianto di rene
L’insufficienza renale cronica si associa di per sé a importanti alterazioni del metabolismo osseo e minerale. Prima del trapianto sono di solito presenti iperfosfatemia, che stimola la secrezione di PTH ed FGF-23, e una riduzione del metabolita attivo della vitamina D (calcitriolo), che causa a sua volta ipocalcemia e stimolazione della secrezione di PTH fino alla comparsa di iperplasia nodulare e autonomizzazione funzionale (iperparatiroidismo terziario). Questi meccanismi omeostatici determinano nelle fasi avanzate pre-trapianto una grave alterazione del metabolismo osseo, denominata osteodistrofia renale, caratterizzata da osteomalacia ed osteoporosi con elevato rischio fratturativo. Data la complessità delle lesioni scheletriche in questi soggetti, la DEXA non predice perfettamente il rischio di fratture. La correzione dell’iperfosfatemia e dell’ipovitaminosi D sono cruciali già prima del trapianto.
Dopo il trapianto, le alterazioni del metabolismo osseo e minerale sono dipendenti sia dalla situazione pre-trapianto sia dalla funzionalità renale post-trapianto che dall’azione dei farmaci immunosoppressivi. L’iperparatiroidismo permane in molti pazienti anche dopo il trapianto, soprattutto a causa di un’autonomizzazione della funzione paratiroidea. Con il trapianto può pertanto osservarsi, dopo un’iniziale ipocalcemia, una più tardiva ipercalcemia (permanente nel 50% dei casi), precoce ipofosfatemia (93% nei primi mesi), causata da iniziale marcata iperfosfaturia indotta dagli elevati livelli di FGF-23 e che tende a normalizzarsi dopo circa un anno, inducendo essa stessa un ulteriore difetto di mineralizzazione ossea. Nei pazienti in cui permane iperfosfaturia tardiva post-trapianto, sembra invece essere implicato il PTH. Per tutte queste ragioni, i pazienti trapiantati di rene presentano con maggiore probabilità una perdita di massa ossea rispetto a quanto si osserva nei pazienti trapiantati con altri organi. L’ipercalcemia post-trapianto è di solito causata da una persistente elevazione del PTH; se il PTH è nel range di norma, vanno invece considerati i potenziali eventi avversi delle terapie immunosoppressive, con possibile ipercalcemia da neoplasie o da infezioni opportunistiche con produzione ectopica di vitamina D (Pneumocystis Jirovecii Pneumonia). La prevalenza di osteoporosi post-trapianto varia tra 11-56%, caratterizzata sia da fratture vertebrali (3-29%) che periferiche (11-43%); la perdita di massa ossea è più marcata nei primi 6 mesi, per continuare più lentamente nei successivi 6-12 mesi. Le alterazioni anatomo-patologiche dell'osso sembrano essere maggiormente dovute alla dose cumulativa di glucocorticoidi e all'iperparatiroidismo terziario. Anche in età pediatrica, dopo il trapianto c’è un aumentato rischio di disordini ossei, quali fratture, necrosi avascolari, dolore osseo e alterazioni della crescita; fattori favorenti l’osteopenia post-trapianto in età pediatrica sono l’osteodistrofia renale pre-esistente, la terapia con glucocorticoidi, il ritardo di crescita e puberale, l’infiammazione cronica e la funzione renale post-trapianto, oltre che una diminuita attività fisica.
In generale, dopo trapianto di rene viene raccomandato uno stretto monitoraggio di calcemia, fosforemia, PTH, DEXA annuale e aggiustamento della terapia immunosoppressiva verso i dosaggi minimi (soprattutto dei glucocorticoidi). L’utilizzo dei bisfosfonati in questa specifica popolazione è generalmente consigliato nel primo anno post-trapianto, ma è ancora oggetto di studio nel lungo termine date le peculiari alterazioni ossee di questa condizione. La terapia con cinacalcet (calciomimetico) si è dimostrata utile sia pre- che post-trapianto nel controllare l’ipercalcemia, anche se non produce un significativo miglioramento della BMD. Non è noto per quanto tempo la terapia con cinacalcet debba o possa essere continuata, anche perché la condizione di iperparatiroidismo post-trapianto può perdurare per molti anni. La paratiroidectomia totale o subtotale (con reimpianto delle paratiroidi) è un’altra opzione terapeutica, soprattutto nei pazienti con persistente ipercalcemia, fratture o ipercalciuria. In caso di paratiroidectomia pre-trapianto, è consigliabile procrastinare il trapianto renale in attesa di una normalizzazione della calcemia e della fosfatemia, a causa della prevedibile ipocalcemia da hungry bone syndrome (“sindrome dell’osso affamato”), che si sommerebbe all’ipocalcemia precoce post-trapianto.
In molti pazienti a 6 mesi post-trapianto renale si verificano aumento di peso, alterazioni del metabolismo glucidico e ipertensione arteriosa, con sviluppo di franca sindrome metabolica. L’aumentato rischio di sindrome metabolica diminuisce al diminuire del tempo in cui si riduce la dose di glucocorticoidi e di inibitori della calcineurina.
A livello tiroideo si descrivono noduli (più frequentemente nelle donne), “low T3 syndrome”, ipotiroidismo subclinico, diminuzione del TSH e dei valori di TBG verosimilmente dovuti all'influenza delle terapie immunosoppressive; l'ipertiroidismo è estremamente raro.
Sono descritti rari casi di diabete insipido idiopatico, che, mascherati dalla presenza di insufficienza renale, si slatentizzano dopo il trapianto renale.
L’uremia ha un impatto negativo anche sulla fertilità, cosa rilevante per i riceventi pediatrici con trapianto fallito. Generalmente negli stati di insufficienza renale cronica maschile vi è uno stato di infertilità e di disfunzione sessuale non pienamente corretti dall'emodialisi o dalla dialisi peritoneale, causati da una compromissione dell'asse ipofisi-gonadi. Tra gli effetti positivi del trapianto di rene vi sarebbe il miglioramento di queste problematiche; dall'altro lato, però, la funzionalità gonadica di questi soggetti potrebbe risultare compromessa in seguito a terapia con inibitori di mTOR.
Riceventi con GFR stimata post-trapianto < 60 mL/min/1.73 m2 a un mese dal trapianto hanno una riduzione della velocità di crescita; si è visto inoltre che i pazienti sottoposti a trapianto prima della pubertà e svezzati dagli steroidi entro 4-6 mesi, raggiungono nel 94% la normale altezza adulta rispetto a quelli trapiantati dopo e con un uguale tempo di svezzamento. Nella maggior parte dei riceventi lo sviluppo puberale è normale: i trapiantati prima dei 5 anni raggiungono la pubertà prima di quelli trapiantati dopo i 5 anni. Riguardo agli effetti degli immunosoppressivi:
- i glucocorticoidi diminuiscono la velocità di crescita;
- tacrolimus e ciclosporina non hanno effetto diretto sulla crescita, ma sono nefrotossici e la diminuita funzione renale è associata a ritardo di crescita;
- sirolimus ha proprietà anti-proliferative e diminuisce la velocità di crescita.
Comunque non sono state rilevate differenze staturali tra pazienti trattati con sirolimus/tacrolimus/everolimus a basse dosi/ciclosporina a basse dosi e pazienti trattati con micofenolato mofetil e ciclosporina a dosi standard. Nei pazienti pediatrici con insufficienza renale cronica e deficit staturale può essere presa in considerazione anche la terapia con rhGH.
Trapianto di fegato
Nell’insufficienza epatica è frequentemente presente ipogonadismo, che nell’uomo si associa a disfunzione erettile, oligospermia, atrofia testicolare, ginecomastia e distribuzione ginoide del tessuto adiposo. L’ipogonadismo è causato da riduzione di testosterone, elevazione di SHBG e iperestrogenemia. I livelli di LH ed FSH si mantengono inappropriatamente bassi, mentre la PRL risulta elevata. Tutte queste alterazioni sono reversibili dopo trapianto di fegato. Normalmente i pazienti con insufficienza epatica terminale hanno bassi livelli di fT3 e fT4, che si normalizzano dopo il trapianto.
Il fegato svolge un’azione di rilievo sull’asse GH/IGF-I. I pazienti con insufficienza epatica presentano elevati livelli di GH basale (resistenza periferica) con bassi livelli di IGF-I, IGF-II e IGF-BPs. Dopo trapianto il quadro si normalizza.
Dopo trapianto, nel 30-35% dei casi si verificano osteopenia o osteoporosi; il rischio è maggiore nei primi 6-12 mesi dopo il trapianto. I fattori di rischio sono terapia con glucocorticoidi, malnutrizione, deficienza di vitamina D con iperparatiroidismo secondario, bassi livelli di IGF-I, colestasi cronica, cirrosi biliare primitiva, immobilità, fumo di sigaretta, sarcopenia, ipogonadismo, alcol.
In molti pazienti a 6 mesi post-trapianto epatico si verificano aumento di peso, alterazioni del metabolismo glucidico, dislipidemia e ipertensione arteriosa con sviluppo di sindrome metabolica: i principali fattori di rischio sono cirrosi epatica su base virale o alcolica, infezioni post-trapianto, terapia con glucocorticoidi, terapia con inibitori della calcineurina e sirolimus, diabete pre-trapianto ed elevato BMI.
In generale, vi è un aumentato rischio di neoplasie maligne post-trapianto. Le neoplasie endocrine post-trapianto sono molto rare: includono il glucagonoma, i tumori neuroendocrini a piccole cellule del piccolo intestino ed il feocromocitoma.
Trapianto di cuore
I pazienti con insufficienza cardiaca cronica in attesa di trapianto presentano alcuni fattori di rischio che possono contribuire alla perdita di massa ossea: insufficienza renale cronica, ipovitaminosi D ed iperparatiroidismo, picco di massa ossea insufficiente (nelle cardiopatie congenite), esposizione prolungata a diuretici dell’ansa o eparina.
Dopo il trapianto è presente osteoporosi a lungo termine nel 20-30% dei casi; la perdita di massa ossea si verifica maggiormente nel primo anno. Si registrano bassi livelli di testosterone, con un intervallo di tempo variabile dopo il trapianto. Nella maggior parte dei casi i bassi livelli di testosterone sono associati a bassi livelli di LH e FSH, suggerendo che il meccanismo sia legato alla soppressione dell'asse ipotalamo-ipofisario collegato ad alte dosi di prednisone, al recente intervento di chirurgia maggiore o all'azione diretta della ciclosporina; in una minoranza di casi le gonadotropine sono elevate, suggerendo un danno gonadico diretto. Di solito l'ipogonadismo si risolve nei 6 mesi successivi al trapianto, ma in alcuni casi può persistere anche a distanza di 2 anni.
Trapianto di polmone
I pazienti in attesa di trapianto potrebbero essere maggiormente soggetti a osteopenia, osteoporosi (fino al 73% dei casi) e fratture vertebrali a causa dei seguenti fattori favorenti: ipercapnia, fumo di sigaretta, terapia cronica con glucocorticoidi, scarsa attività fisica. Nei pazienti con fibrosi cistica possono intervenire anche: insufficienza pancreatica (che riduce l’assorbimento di calcio e vitamina D), ipogonadismo, ridotto picco di massa ossea.
Trapianto di cellule staminali ematopoietiche
Il trapianto di cellule staminali ematopoietiche è preceduto da un’elevata dose di radiazione e di chemioterapici, compresi gli agenti alchilanti. Il danno a carico dell’apparato riproduttivo dipende dalla dose totale di radiazione, dal suo frazionamento, dal fatto che vi sia stato o meno un trattamento radioterapico aggiuntivo a livello cerebrale o gonadico, dall’età del paziente in relazione allo sviluppo puberale e dal sesso:
- nelle donne l'insufficienza ovarica post auto- o allo-trapianto è una complicanza frequente con remota possibilità di recupero. Altra causa è l'uso di un analogo del GnRH, soppressore dell'asse GnRH-Gn-ovaio, impiegato per prevenire le emorragie vaginali post-trapianto. Il danno ovarico è stato anche dimostrato in corso di GVHD. La capacità dell’analogo del GnRH di proteggere la funzione ovarica dopo il trapianto è controversa;
- negli uomini, sia la chemioterapia che la radioterapia (> 2000 cGy) hanno un effetto tossico sulle cellule dell'epitelio germinale (maggiormente) ed in quelle del Leydig, con conseguente ipogonadismo primario con rara possibilità di recupero funzionale. Il danno sull’epitelio germinale è variabile in relazione alla dose dei farmaci e alla loro combinazione (peggiorativo se busulfano + ciclofosfamide). Tra i fattori predittivi di residua spermatogenesi ricordiamo la giovane età e l’assenza di GVHD. La maggior parte dei pazienti allo- e auto-trapiantati, a causa delle problematiche precedenti, non sono fertili.
Si è visto che alcuni fattori favoriscono il deficit di GH post-trapianto: età pediatrica, radioterapia (al posto della chemioterapia), singola dose di irradiazione corporea (al posto della dose frazionata) e la chemioterapia intra-tecale. Altro fattore implicato è la GHVD (a sua volta associata a terapia cronica con glucocorticoidi), responsabile di un danno multiorgano, che coinvolge dunque anche l'ipofisi. La radioterapia influenza la crescita lineare attraverso una condizione di GH deficit e displasia scheletrica (quest’ultima può comportare una maggiore riduzione della crescita del tronco rispetto agli arti). Ricordiamo comunque che alcuni pazienti con diagnosi di GHD in età pediatrica possono presentare una normale riserva di GH quando vengono ritestati in età adulta. La terapia sostitutiva con rhGH è indicata nei pazienti con deficit ormonale documentato, in remissione da un anno, e che mostrano una compromissione della crescita staturale.
Per quanto riguarda la tiroide, dopo allotrapianto e, molto raramente, dopo autotrapianto sono descritti low T3 syndrome, tiroidite cronica, ipo e iper-tiroidismo sub-clinici, e trasformazione neoplastica (carcinoma papillifero e follicolare della tiroide). Ipotiroidismi permanenti o transitori sono stati descritti anche dopo anni dall'irradiazione totale corporea e dopo preparazione con busulfano e ciclofosfamide.
La soppressione dell’asse ipotalamo-ipofisi-surrene è maggiormente dovuta alle alte dosi di glucocorticoidi utilizzate dopo il trapianto, che sopprimono la produzione di CRH e ACTH; di solito il deficit è transitorio. Recentemente sono stati anche descritti casi di insufficienza surrenalica post-irradiazione totale corporea.
La prevalenza di osteopenia è vicina al 50% a distanza di 4-6 anni dal trapianto di midollo. La perdita di massa ossea è maggiore nel primo anno post-trapianto. Tra i fattori di rischio ricordiamo la terapia con glucocorticoidi a causa della GVHD, la pregressa radioterapia (indice di GHD) e il deficit della funzione gonadica. Un altro fattore favorente è determinato dalla riduzione delle cellule osteoprogenitrici di origine midollare. Sono state descritte anche necrosi avascolare e osteomalacia. Questi pazienti dovrebbero essere monitorati con la DEXA, e trattati con misure preventive (supplementi di calcio e vitamina D, cessazione del fumo o calo ponderale), farmaci (bisfosfonati) e correzione dell’ipogonadismo.
Dopo trapianto di midollo è stata infine descritta insulino-resistenza e sindrome metabolica. L’irradiazione corporea globale si può associare a diabete, sia con un meccanismo di insulino-resistenza sia per una insulinopenia.
Bibliografia essenziale
- Taweesedt PT, Disthabanchong S. Mineral and bone disorder after kidney transplantation. World J Transplant 2015, 5: 231-42.
- Issa DH, Alkhouri N. Long-term management of liver transplantation recipients: a review for the internist. Cleve Clin J Med 2015, 82: 361-72.
- Shivaswamy V, Boerner B, Larsen J. Post-transplant diabetes mellitus: causes, treatment, and impact on outcomes. Endocr Rev 2016, 37: 37-61.
- Shalet SM, Didl M, Oglivy-Stuart Al, et al. Growth and endocrine function after bone marrow transplantation. Clin Endocrinol 1995, 42: 333-9.
- Subramanian S, Trence DL. Immunosuppressive agents: effects on glucose and lipid metabolism. Endocrinol Metab Clin North Am 2007, 36: 891-905.
- Chemaitilly W, Sklar CA. Endocrine complications of hematopoietic stem cell transplantation. Endocrinol Metab Clin North Am 2007, 36: 983-98.
- Maalouf NM, Shane E. Osteoporosis after solid organ transplantation. J Clin Endocrinol Metab 2004, 90: 2456-65.
Manifestazioni psichiche in corso di malattie endocrine
Pierantonio Conton
UOC Medicina Interna - UOSD Malattie Endocrine, del Metabolismo e Nutrizione, Ospedale Dell'Angelo, ASL 12 Veneziana
Tireotossicosi
Le alterazioni psicologiche e comportamentali dovute alla tireotossicosi sono molteplici e con incidenza variabile, che può raggiungere comunque il 60-70% dei pazienti.
Classicamente è riportata una condizione di ansia e disforia, labilità emotiva, insonnia e, talora, disturbi dell’intelletto. La capacità di concentrazione può essere particolarmente compromessa. D’altra parte, questo può essere uno dei disturbi più precoci della tireotossicosi, assieme all’irrequietezza e ai tremori. Il paziente appare irritabile, nervoso, facilmente propenso alla collera; talora esprime idee di riferimento o franca paranoia. I pensieri e l’eloquio possono fluire velocemente e talvolta susseguirsi sconnessi. L’attività motoria è accelerata e associata ad agitazione. I disturbi del sonno includono, oltre all’insonnia, sogni vividi e incubi, portando a conseguente senso di stanchezza diurna (elemento distintivo dal vero disturbo maniacale). Più raramente, in alcuni pazienti, le alterazioni comportamentali possono progredire verso forme aspecifiche di psicosi più strutturate, con sentimenti di delusione o di natura paranoide. Convulsioni epilettiformi sono rare. Nella “tempesta tireotossica ” l’insorgenza dei disturbi può essere molto rapida.
In contrasto con questo quadro clinico, alcuni pazienti denotano uno stato psichico definibile come “tireotossicosi apatica”, che mima una sorta di depressione, più comunemente osservabile nel paziente anziano, raramente tuttavia anche nell’adolescente. Questa sindrome si caratterizza per apatia, letargia, pseudo-demenza, calo ponderale, deflessione del tono dell’umore a esordio “melancolico”. Tale corteo sintomatologico può talora ritardare la diagnosi di tireotossicosi o, peggio, condurre a terapie farmacologiche psichiatriche potenzialmente esacerbanti la patologia tiroidea (1-3).
Ipotiroidismo
È ben noto il quadro neurologico con grave ritardo mentale (“cretinismo”) che si associa all’ipotiroidismo neonatale. Nell’ipotiroidismo dell'adulto gli iniziali cambiamenti del comportamento e le alterazioni neuro-psicologiche sono sfumati, aspecifici e mal definiti dal paziente stesso (ad es. stanchezza, facile affaticabilità), con conseguenti difficoltà o ritardi diagnostici. I disturbi cognitivi comprendono scarsa attenzione e capacità di concentrazione, rallentamento del pensiero, difficoltà nel calcolo e nella comprensione di quesiti complessi. Risulta frequentemente ridotta la capacità della memoria a breve termine (più raramente la memoria remota), come anche, in fasi più avanzate, l’abilità nello svolgere le comuni attività della routine quotidiana, fino ad arrivare a forme di demenza, reversibili, specie nel paziente anziano. Appare ridotta sia la reattività agli stimoli esterni e l’interesse per il mondo circostante, che la capacità di portare a termine l’attività lavorativa. La libido è diminuita. L’eloquio risulta più lento, “impoverito”, talvolta con ripetizioni. L’attività motoria è rallentata. In alcuni casi si realizza una sindrome melancolica, con facilità al pianto e alla delusione, stipsi, insonnia, idee suicidarie. Negli stadi più avanzati dell’ipotiroidismo possono comparire sindromi allucinatorie visive, comportamento bizzarro e idee paranoidi, nonché, nelle condizioni più gravi, sonnolenza, letargia: i pazienti possono trascorrere molte ore diurne nel sonno, fino ad arrivare allo stato soporoso o al coma.
Da segnalare inoltre una specifica forma di encefalopatia nella tiroidite di Hashimoto, caratterizzata da remissioni e recidive, crisi comiziali focali o generalizzate, transitori deficit neurologici e una molteplicità di manifestazioni psichiatriche, comprendenti allucinazioni, demenza e psicosi acuta (1-3).
Ipercortisolismo
Le alterazioni dell’asse ipotalamo-ipofisi-surrene (HPA), sia endogene che conseguenti all’impiego di farmaci che ne alterano i meccanismi di feed-back (tipicamente i “cortisonici”), si correlano allo sviluppo di alterazioni comportamentali, fino allo sviluppo di psicopatie, in relazione soprattutto alla cospicua presenza nel sistema nervoso centrale di recettori per i glucocorticoidi (GR). La sindrome di Cushing si accompagna spesso inizialmente a disfunzione affettiva; in fase conclamata si osservano disturbi maniacali, sindromi ansiose, disfunzione cognitiva e ideazione suicidaria presente nel 10% dei pazienti. La depressione è comunque il disturbo psichiatrico più frequentemente associato: si tratta di una forma atipica (con livelli di CRH soppressi), caratterizzata da iperfagia, astenia ed eccessiva sonnolenza, che si contrappone alla forma melancolica presente nell’iperattività dell’asse HPA sostenuta dallo stress (iperarousal, insonnia, anoressia). Studi più recenti hanno dimostrato l’esistenza di alterazioni funzionali dei GR nel SNC in alcune forme di depressione (“resistenza ai glucorticoidi”), in cui l’ipercortisolemia risulta essere un epifenomeno di compensazione e non secondario all’attivazione dell’asse HPA. In considerazione del loro largo impiego clinico, si deve segnalare che anche la somministrazione di glucocorticoidi sintetici, anche per brevi periodi di tempo, è potenzialmente in grado di causare disturbi quali mania/ipomania (più frequenti), inoltre apatia, ansia, disturbi da attacco di panico, depersonalizzazione, delirium. L’assunzione cronica è più frequentemente associata allo sviluppo di depressione simile alla sindrome di Cushing (2-6).
Iposurrenalismo
Nel morbo di Addison si riscontrano frequentemente deficit di concentrazione, irrequietezza, irritabilità, disturbi del sonno. I disturbi elettrolitici secondari in fase di scompenso possono condurre ad alterazioni della memoria e dello stato di coscienza, fino al coma. Nel 20% dei casi si associa depressione, con apatia, astenia, perdita dell’iniziativa. Nel 20-40% dei pazienti si riscontrano forme di psicosi, con allucinazioni, deliri paranoidi, negativismo, postura bizzarra o catatonica. Possono infine associarsi dispercezioni sensoriali. In una minoranza di casi tali disturbi possono persistere anche dopo l’inizio della terapia sostitutiva (2-6).
Acromegalia
I dati presenti in letteratura, talora non univoci, riportano come principale alterazione nell'acromegalia la perdita di iniziativa e spontaneità, con instabilità dell’umore. Sono presenti inoltre alterazioni dell’autostima, dismorfismo della percezione corporea, talora segni di ansia e rabbia nei confronti dell’inefficienza medica. Non sono classicamente riportate evidenti alterazioni intellettuali. Schizofrenia e psicosi maniaco-depressiva sono eventi rari. Le alterazioni neuropsichiatriche non appaiono correlabili ai valori plasmatici di GH, d‘altra parte l’impiego di analoghi della somatostatina sembra migliorare la qualità di vita psicologica e di relazione dei pazienti acromegalici (2,3).
Deficit di GH
Nell’infanzia il deficit di ormone somatotropo è correlabile a ritardo dell’apprendimento e dell’attenzione, ridotto rendimento scolastico. I bambini si dimostrano spesso psicologicamente immaturi e talora con disturbi della personalità. Nei pazienti adulti si riscontrano più spesso astenia, ridotta qualità del sonno, alte percentuali di disturbi d’ansia, fobia, ritiro sociale e depressione, apparentemente reversibili con la terapia sostitutiva. Sono descritte correlazioni positive tra concentrazioni plasmatiche di IGF-1, quoziente intellettivo e livello educazionale, tanto più forti quanto più precocemente insorge il deficit. Relazioni tra disponibilità di GH a livello ippocampale e turnover della dopamina sembrano spiegare possibili deficit della memoria (2,3).
Iperprolattinemia
Nell'iperprolattinemia, più spesso associati al sesso femminile, si riscontrano stati di depressione, ansia, ostilità. Il 30% delle pazienti presenta criteri diagnostici di depressione maggiore. Nel sesso maschile il calo della libido classicamente riportato può talora condurre a stati depressivi. Attraverso le vie endorfiniche l’eccesso di PRL aumenta la soglia del dolore e l’appetito. I disturbi migliorano con il trattamento dopaminergico. Le strette relazioni circolari tra prolattina, emozioni e sentimenti, suggeriscono che alterazioni della personalità e influenze comportamentali - ambientali potrebbero avere un ruolo nella stessa genesi della patologia iperprolattinemica. La PRL è identificata come componente di una serie di comportamenti e meccanismi metabolici necessari per la cura della prole (“programma materno”) (1,3,7).
Ormoni sessuali
Rapide oscillazioni nei livelli di estrogeni, come ad esempio nel peri-partum, possono ricoprire un ruolo nell’insorgenza di disturbi bipolari e depressione maggiore nel sesso femminile. La loro progressiva riduzione nella fase peri-menopausale è associata a un aumento di incidenza di depressione. Nel maschio il deficit di testosterone può associarsi, oltre al calo della libido, a deflessione del tono dell’umore, irritabilità, deficit della concentrazione e della memoria a breve termine, peggioramento della performance lavorativa. L’impiego di steroidi anabolizzanti e androgeni negli atleti a scopo dopante è associato a disturbi comportamentali e dell’umore, ansia, aggressività, disturbi del comportamento alimentare e sviluppo di psicosi croniche in individui predisposti, correlabili all’entità e al prolungato periodo di assunzione. Brusche sospensioni di tali pratiche possono aumentare l’incidenza di depressione e suicidio (1,8,9).
Bibliografia
- Werner SC, Ingbar SH, et al. The Thyroid. A Fundamental and clinical text, 2005 (ISBN 0-7817-5047-4).
- De Groot L, et al. Endocrinology, 5th Edition, 2006 (ISBN 0-7216-0376-9).
- Mondelli V, et al. I disturbi psichiatrici dei pazienti con malattie endocrine. Riv Psichiatr 2006, 41: 10-5.
- Juruena MF, et al. The hypotalamic pituitary adrenal axis, glucocorticoid receptor function and relevance to depression. Rev Bras Psiquiatr 2004, 26, 189-201.
- Pariante CM. Risk factors for development of depression and psychosis: glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci 2009, 1179: 144-52.
- Kenna HA, et al. Psychiatric complications of treatment with corticosteroids: review with case report. Psychiatry Clin Neurosci 2011, 65: 549-60.
- Ali S, et al. Management of psychosis associated with a prolactinoma: case report and review of the literature. Psychosomatics 2010, 51: 370-6.
- Meinhard N, et al. The role of estrogen in bipolar disorder, a review. Nord J Psychiatry 2014, 68: 81-7.
- Piacentino D, et al. Anabolic-androgenic steroid use and psycopathology in athletes. A systematic review. Curr Neuropharmacol 2015, 13: 101-21.
Conseguenze endocrine delle malattie psichiatriche
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
(aggiornato all’8 luglio 2020)
TIROIDE E PSICHIATRIA
EFFETTI DEGLI ORMONI TIROIDEI SUL SNC
Gli ormoni tiroidei giocano un ruolo fondamentale nell'omeostasi metabolica, nella crescita e sviluppo, soprattutto del sistema nervoso centrale (SNC), che appare molto sensibile alla loro azione durante la vita intra-uterina.
La tiroide fetale nell’uomo diventa pienamente funzionante a partire dal II trimestre di gestazione, ma il feto esprime recettori per gli ormoni tiroidei dalla 9° settimana, poiché da questo momento gli ormoni tiroidei di origine materna regolano il processo di proliferazione e migrazione neuronale nella corteccia cerebrale, ippocampo ed eminenza mediale del feto. Dalla 14° settimana anche la tiroide fetale contribuisce alla presenza di ormoni tiroidei. In questa fase cominciano la neurogenesi, la migrazione neuronale, la crescita assonale, l’arborizzazione dei dendriti, la formazione delle sinapsi, la differenziazione delle cellule gliali e la formazione della mielina (1). Dalla 28° settimana la madre e il feto contribuiscono in egual misura allo sviluppo del SNC, ma poiché la tiroide fetale non è pienamente matura fino alla nascita, una condizione di insufficiente produzione di ormoni materni può indurre effetti avversi anche in questa fase (2).
Dopo la nascita il completamento di alcuni processi (mielinizzazione e migrazione delle cellule granulari nel giro dentato dell’ippocampo e del cervelletto, delle cellule piramidali nella corteccia cerebrale e delle cellule di Purkinje nel cervelletto) dipenderà solo dall'attività della tiroide infantile (1).
Alla luce di queste evidenze, una condizione di franco ipotiroidismo materno, soprattutto nel I trimestre di gravidanza, indurrà la comparsa di danni irreversibili del SNC (tabella) (3), che clinicamente si manifesteranno con ritardo dello sviluppo cognitivo, strabismo, sordità e disturbi dell’eloquio, spasticità o paralisi degli arti inferiori, atassia, fino al cosiddetto cretinismo (3). Alcuni studi in pazienti nati da madri con disfunzione tiroidea sembrano inoltre indicare un aumentato rischio di epilessia, disordini dello spettro autistico, disordini da deficit di attenzione/iperattività (ADHD) e altre condizioni psichiatriche (4,5).
| Alterazioni del SNC secondarie a deficit di ormoni tiroidei (in ordine cronologico , dal I trimestre alla vita post-natale) |
| Diminuzione della proliferazione delle cellule staminali |
| Deficit di migrazione neuronale |
| Ritardo della proliferazione neuronale |
| Diminuita espressione dei fattori di differenziazione neuronale |
| Deficit nell’inizio dell’attività primitiva di rete |
| Riduzione dello spessore corticale |
| Displasia corticale |
| Anomalie nella deposizione e stratificazione della corteccia cerebellare |
| Deficit di sviluppo di dendriti e assoni |
| Diminuita espressione delle proteine coinvolte nella plasticità sinaptica |
| Ritardo di mielinizzazione e ridotta guida assonale |
L’ipotiroidismo subclinico materno così come l’ipotiroidismo congenito (atireosi) sembrano associati a minor quoziente intellettivo (QI) nei bambini in età scolare, soprattutto nelle scale di performance (6-8), sebbene i dati della letteratura non siano sempre concordi. Per quanto riguarda l’azione degli ormoni tiroidei sul cervello adulto, sono ormai numerosi gli studi condotti in vitro e in vivo, sugli animali e sull’uomo, che dimostrano come questi ormoni esercitino un ruolo fondamentale nella neurogenesi, sia a livello della zona sub-capsulare dell’ippocampo che nella zona sub-ventricolare che delimita il III ventricolo, cioè nella formazione di nuovi neuroni maturi o di interneuroni e neuroni peri-glomerulari che vanno a integrarsi nel circuito olfattorio (9).
FUNZIONE TIROIDEA E DISTURBI PSICHIATRICI
Depressione
Ipotiroidismo franco: rallentamento del pensiero e del linguaggio, riduzione dell'attenzione e apatia sono tutti segni/sintomi di una condizione di ipotiroidismo conclamato, che possono essere confusi con la depressione (10). L’esecuzione di test può svelare un incremento dei punteggi relativi ad ansia e depressione, che possono migliorare, ma non sempre completamente, con la terapia sostitutiva (11). Raramente il paziente ipotiroideo può sviluppare un quadro di agitazione e franca psicosi (12).
Ipotiroidismo subclinico: il link con la depressione è fortemente controverso. Alcuni dati sembrano indicare una maggiore prevalenza di ipotiroidismo subclinico nei pazienti affetti da depressione (10,13) e almeno dagli anni ’60 è invalso l’uso di aggiungere ormoni tiroidei alla terapia anti-depressiva, anche a dosi sovra-fisiologiche (14), per potenziarne l’effetto e accelerare il miglioramento clinico nei non responder, sebbene una più recente meta-analisi degli studi sull’associazione SSRI + T3 non abbia dimostrato una risposta clinica più rapida rispetto alla terapia con soli SSRI (15).
Nella pratica clinica, quindi, i pazienti affetti da depressione o riduzione del tono dell’umore vengono generalmente sottoposti a valutazione della funzione tiroidea ed eventualmente trattati con L-tiroxina se presente una condizione anche modesta di ipotiroidismo subclinico, nella speranza che tale terapia determini un miglioramento dei disturbi affettivi (16). Un recente lavoro retrospettivo condotto in UK su più di 50.000 pazienti ha, infatti, evidenziato un progressivo incremento durante l’osservazione della prescrizione e del dosaggio di L-T4, anche per condizioni di modesto ipotiroidismo subclinico e in quei pazienti nei quali gli effetti collaterali avrebbero potuto essere maggiori dei benefici, suggerendo un uso che va oltre il semplice controllo del TSH. Infatti, i pazienti con astenia o depressione all’inizio dello studio venivano trattati con dosaggi più elevati di L-T4 (17). Studi più recenti non hanno dimostrato però né una maggiore prevalenza di depressione nei pazienti con ipotiroidismo subclinico rispetto agli eutiroidei (18,19), né che il trattamento con L-T4 in questa classe di pazienti determini un miglioramento dei punteggi di depressione (20-22).
Nel 2018 è stato pubblicato uno studio prospettico (23) su oltre 90.000 adulti (39 ± 6.7 anni) valutati per funzionalità tiroidea, avendo come endpoint lo sviluppo di sintomi depressivi in un periodo medio di follow-up di due anni. Lo studio non ha dimostrato alcuna associazione tra ipotiroidismo subclinico e sintomi depressivi incidenti. Il risultato rimaneva simile nelle diverse classi di età e sesso e non era influenzato dall’aggiustamento per potenziali fattori confondenti (BMI, fumo, uso di alcool, esercizio fisico, comorbilità).
Alcuni piccoli studi sembrano dimostrare invece che l’ipotiroidismo subclinico sia in grado di modificare, seppure sottilmente, alcuni domini cognitivi (memoria verbale e spaziale, funzioni esecutive) e si associ a anomalie delle aree frontali nella MR funzionale. Alcuni soggetti trattati con L-T4 per 6 mesi hanno presentato un miglioramento radiologico e dei punteggi cognitivi (24,25). Molti pazienti ipotiroidei in trattamento con L-T4 continuano a riferire un disturbo dell’umore o delle funzioni cognitive, nonostante valori normali di TSH. Alcuni studi sembrano indicare che in questi pazienti i disturbi affettivi siano legati alla consapevolezza della malattia tiroidea e alla necessità di assumere la terapia, piuttosto che alla funzione tiroidea stessa (26-28). Un’altra possibile spiegazione è la presenza di polimorfismi della desiodasi 2 o dei trasportatori degli ormoni tiroidei, che possono teoricamente indurre una condizione di ipotiroidismo intra-cellulare con bassi livelli di T3 (29,30). Tuttavia, i dati non sembrano dimostrare una correlazione tra depressione e livelli di T3 negli ipotiroidei trattati (31), né che la terapia combinata LT4 + LT3 dia beneficio sulle funzioni cognitive o sull’umore (32,33).
Alla luce di tutte queste evidenze, i disturbi affettivi non sembrano associati all’ipotiroidismo subclinico, ma rappresentano una condizione psichiatrica indipendente molto comune nella popolazione generale e che si presenta con uguale frequenza nei pazienti con ipotiroidismo subclinico. Spesso questi sintomi sono più evidenti in quei pazienti più preoccupati per la propria condizione endocrinologica, sebbene non possa essere completamente esclusa la presenza di sottili deficit dell’umore in particolari sottogruppi di pazienti.
2b. Schizofrenia e disordini bipolari
Diversi studi epidemiologici sostengono la presenza di un legame tra disturbi psichiatrici e malattie autoimmuni, con un aumento del rischio di schizofrenia e disordini bipolari negli individui che hanno una storia personale o familiare di malattie autoimmuni (34 35). La presenza di autoimmunità tiroidea è stata anche proposta come fattore di rischio indipendente per lo sviluppo di disturbi bipolari, senza associazione con l’esposizione al litio (36). A loro volta i pazienti con disturbi bipolari sembrano presentare un maggiore rischio di sviluppare tiroiditi autoimmuni e alcuni dati della letteratura sembrano suggerire che in alcune famiglie gli AbTPO e la patologia psichiatrica vengano trasmessi in modo associato, tanto da ipotizzare che i livelli di AbTPO possano essere utilizzati come possibile endofenotipo per i disordini bipolari stessi (37). Sia l’iper- che l’ipotiroidismo autoimmune (Graves e Hashimoto) possono essere riscontrati nei pazienti bipolari e schizofrenici, tuttavia l’ipertiroidismo sembra preponderante nei primi mentre nella schizofrenia sembra maggiore il rischio di ipotiroidismo (38). Tale dato sembra confermato anche da un recente studio di popolazione condotto in Israele (39), che ha coinvolto più di 40.000 ipotiroidei e 40.000 controlli, e che ha dimostrato come la schizofrenia sia associata in modo indipendente all’ipotiroidismo, anche dopo correzione per sesso e altri fattori quali età, fumo e classe sociale. Tali evidenze suggeriscono la necessità di valutare sempre la funzionalità tiroidea nei pazienti psichiatrici.
BIBLIOGRAFIA
- Williams GR. Neurodevelopmental and neurophysiological actions of thyroid hormone. J Neuroendocrinol 2008, 20: 784-94.
- Trueba SS, Augè J, Mattei G, et al. PAX8, TITF1, and FOXE1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis- associated malformations. J Clin Endocrinol Metab 2005, 90: 455-62.
- Prezioso G, Giannini C, Chiarelli F. Effect of thyroid hormones on neurons and neurodevelopment. Horm Res Paediatr 2018, 90: 73–81.
- Andersen SL, Laurberg P, Wu CS, Olsen J. Maternal thyroid dysfunction and risk of seizure in the child: a Danish nationwide cohort study. J Pregnancy 2013, 2013: 636705.
- Andersen SL, Olsen J, Wu CS, Laurberg P. Psychiatric disease in late adolescence and young adulthood. Foetal programming by maternal hypothyroidism? Clin Endocrinol (Oxf) 2014, 81: 126–33.
- Korevaar TIM, Muetzel R, Medici M, et al. Association of maternal thyroid function during early pregnancy with offspring IQ and brain morphology in childhood: a population-based prospective cohort study. Lancet Diabetes Endocrinol 2016, 4: 35-43.
- Haddow JE, Palomaki GE, Allan WC, et al. Maternal thyroid deficiency during pregnancy and subsequent neuropsychological development of the child. N Engl J Med 1999, 341: 549-55.
- Dimitropoulos A, Molinari L, Etter K, et al. Children with congenital hypothyroidism: long-term intellectual outcome after early high-dose treatment. Pediatr Res 2009, 65: 242–8.
- Fanibunda SE, Desouza La, Kapoor R, et al. Thyroid hormone regulation of adult neurogenesis. Vitam Horm 2018, 106: 211-51.
- Samuels MH. Psychiatric and cognitive manifestations of hypothyroidism. Curr Opin Endocrinol Diabetes Obes 2014, 21: 377–83.
- Davis JD, Tremont G. Neuropsychiatric aspects of hypothyroidism and treatment reversibility. Minerva Endocrinol 2007, 32: 49–65.
- Easson WM. Myxedema with psychosis. Arch Gen Psychiatry 1966, 14: 277-83.
- Hage MP, Azar ST. The link between thyroid function and depression. J Thyroid Res 2012, 2012: 590648.
- Baumgartner A. Thyroxine and the treatment of affective disorders: an overview of the results of basic and clinical research. Int J Neuropsychopharmacol 2000, 3: 149–65.
- Papakostas GI, Cooper-Kazaz R, Appelhof BC, et al. Simultaneous initiation (coinitiation) of pharmacotherapy with triiodothyronine and a selective serotonin reuptake inhibitor for major depressive disorder: a quantitative synthesis of double-blind studies. Int Clin Psychopharmacol 2009, 24: 19–25.
- Samuels HM. Subclinical hypothyroidism and depression: is there a link. J Clin Endocrinol Metab 2018, 103: 2061–4.
- Taylor PN, Iqbal A, Minassian C, et al. Falling threshold for treatment of borderline elevated thyrotropin levels-balancing benefits and risks: evidence from a large community-based study. JAMA Intern Med 2014, 174: 32–9.
- Wiersinga WM. Therapy of endocrine disease: T4 + T3 combination therapy: is there a true effect? Eur J Endocrinol 2017, 177: R287–96.
- Blum MR, Wijsman LW, Virgini VS, et al; PROSPER study group. Subclinical thyroid dysfunction and depressive symptoms among the elderly: a prospective cohort study. Neuroendocrinology 2015, 103: 291–9.
- Jorde R, Waterloo K, Storhaug H, et al. Neuropsychological function and symptoms in subjects with subclinical hypothyroidism and the effect of thyroxine treatment. J Clin Endocrinol Metab 2006, 91: 145–53.
- Kong WM, Sheikh MH, Lumb PJ, et al. A 6-month randomized trial of thyroxine treatment in women with mild subclinical hypothyroidism. Am J Med 2002, 112: 348–54.
- Parle J, Roberts L, Wilson S, et al. A randomized controlled trial of the effect of thyroxine replacement on cognitive function in community-living elderly subjects with subclinical hypothyroidism: the Birmingham Elderly Thyroid study. J Clin Endocrinol Metab 2010, 95: 3623–32.
- Kim JS, Zhang Y, Chang Y, et al. Subclinical hypothyroidism and incident depression in young and middle-age adults. J Clin Endocrinol Metab 2018, 103: 1827–33.
- Zhu DF, Wang ZX, Zhang DR, et al. fMRI revealed neural substrate for reversible working memory dysfunction in subclinical hypothyroidism. Brain 2006, 129: 2923–30.
- Yin JJ, Liao LM, Luo DX, et al. Spatial working memory impairment in subclinical hypothyroidism: an fMRI study. Neuroendocrinology 2013, 97: 260–70.
- Panicker V, Evans J, Bjoro T, et al. A paradoxical difference in relationship between anxiety, depression and thyroid function in subjects on and not on T4: findings from the HUNT study. Clin Endocrinol 2009, 71: 574–80.
- Kramer CK, von Muhlen D, Kritz-Silverstein D, et al. Treated hypothyroidism, cognitive function, and depressed mood in old age: the Rancho Bernardo Study. Eur J Endocrinol 2009, 161: 917–21.
- Samuels MH, Kolobova I, Smeraglio A, et al. The effects of levothyroxine replacement or suppressive therapy on health status mood, and cognition. J Clin Endocrinol Metab 2014, 99: 843–51.
- Panicker V, Saravanan P, Vaidya B, et al. Common variation in the DIO2 gene predicts baseline psychological well-being and response to combination thyroxine plus triiodothyronine therapy in hypothyroid patients. J Clin Endocrinol Metab 2009, 94: 1623–9.
- van der Deure WM, Appelhof BC, Peeters RP, et al. Polymorphisms in the brain-specific thyroid hormone transporter OATP1C1 are associated with fatigue and depression in hypothyroid patients. Clin Endocrinol (Oxf) 2008, 69: 804–11.
- Samuels MH, Kolobova I, Smeraglio A, et al. Effect of thyroid function variations within the laboratory reference range on health status, mood, and cognition in levothyroxine-treated subjects. Thyroid 2016, 26: 1173–84.
- Wartofsky L. Combination L-T3 and L-T4 therapy for hypothyroidism. Curr Opin Endocrinol Diabetes Obes 2013, 20: 460–6.
- Wiersinga WM. Paradigm shifts in thyroid hormone replacement therapies for hypothyroidism. Nat Rev Endocrinol 2014, 10: 164–74.
- Arias I, Sorlozano A, Villegas E, et al. Infectious agents associated with schizophrenia: a meta-analysis. Schizophr Res 2012, 136: 128–36.
- Benros ME, Pedersen MG, Rasmussen H, et al. A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am J Psychiatry 2014, 171: 218–26.
- Kupka RW, Nolen WA, Post RM, et al. High rate of autoimmune thyroiditis in bipolar disorder: lack of association with lithium exposure. Biol Psychiatry 2002, 51: 305–11.
- Hasler G, Drevets WC, Manji H K, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology 2004, 29: 1765–81.
- Cremaschi L, Kardell M, Johansson V, et al. Prevalences of autoimmune diseases in schizophrenia, bipolar I and II disorder, and controls. Psychiatry Res 2017, 258: 9–14.
- Sharif K, Tiosano S, Watad A, et al. The link between schizophrenia and hypothyroidism: a population-based study. Immunol Res 2018, 66: 663–7.
CORTICOSTEROIDI E PSICHIATRIA
ASSE HPA E RISPOSTA ALLO STRESS
Il processo di risposta allo stress determina l’attivazione diretta e coordinata di diversi sistemi. L’asse ipotalamo-ipofisi-surreni (HPA) rappresenta un sistema centrale di risposta biologica allo stress e appare essenziale nel permettere all’individuo di contrastare quegli eventi che minacciano la sopravvivenza fisica o i cosiddetti stressor psico-sociali, grazie a un adattamento dinamico di fronte al mutare delle sollecitazioni ambientali (1).
L’attivazione dell’asse HPA si traduce in una rete di complessi segnali strettamente regolati da meccanismi inibitori (feed-back) e permissivi (feed-forward). I neuroni della regione parvo-cellulare del nucleo para-ventricolare dell’ipotalamo rilasciano CRH e vasopressina (AVP) nella circolazione portale attraverso l’eminenza mediana. I due ormoni si legano quindi ai recettori presenti sulle cellule ipofisarie e stimolano la secrezione di ACTH, il quale attraverso il circolo ematico giunge al surrene, dove induce sintesi e rilascio di cortisolo (2). Una volta sintetizzato, il cortisolo è in grado di agire ubiquitariamente e determinare una molteplice serie di effetti, attraverso meccanismi genomici e non-genomici. Gli effetti genomici sono mediati dal suo legame con due recettori specifici: i recettori ad alta affinità per i mineralcorticoidi (MR) e i recettori a bassa affinità per i glucocorticoidi (GR), localizzati all’interno del citoplasma (3,4). In condizioni di secrezione basale e all’inizio di un evento stressante, il cortisolo si lega primariamente ai MR, che mostrano la più elevata affinità, mentre i GR vengono legati progressivamente quando la secrezione di cortisolo aumenta durante la risposta allo stress e al momento del picco di secrezione circadiana (5). Una volta legato al cortisolo, il complesso recettore-cortisolo trasloca all’interno del nucleo, dove si lega a sua volta a determinati siti del DNA (glucocorticoid response elements) situati nella regione promoter di geni target, dei quali modula l’espressione, attivandone o reprimendone la trascrizione (6).
Nella risposta allo stress, gli effetti genomici più lenti sono affiancati da quelli non genomici, mediati da recettori di membrana (3,7). Gli effetti dei meccanismi non genomici sono più veloci e necessari per il rapido contrapporsi al mutare delle situazioni ambientali (8). Attraverso questa via il cortisolo modula l’attività di diversi sistemi di neurotrasmettitori, come serotonina, acido-γ-aminobutirrico tipo A (GABAA), glutamato, dopamina e acetilcolina, con effetti sui processi di gratificazione/ricompensa, attenzione, funzioni esecutive, umore ed emozioni (9,10).
I primi effetti dell’incremento acuto della secrezione di cortisolo sono rappresentati da aumento del rilascio di glucosio, necessario per fornire energia ai diversi organi coinvolti nella risposta allo stress, intensificazione dello stato di vigilanza e modificazione della risposta immune (11). Allo stesso tempo vengono inibite le funzioni non immediatamente necessarie, come crescita e riproduzione (7).
Una volta cessato lo stimolo stressante, entrano in gioco i meccanismi di feed-back negativo, che cercano di riportare il sistema alla situazione di base, limitando l’esposizione dei tessuti, compreso il SNC, al cortisolo (12). Attraverso i GR, il cortisolo stesso inibisce a livello ipotalamico e ipofisario l’ulteriore rilascio di CRH e ACTH, mentre il legame con questi recettori nell’ippocampo e nella corteccia pre-frontale contribuisce all’effetto inibitorio attraverso un impulso GABAergico diretto al nucleo para-ventricolare (13). Un altro meccanismo con il quale viene limitata la disponibilità del cortisolo è rappresentato dal suo legame con la cortisol-binding-protein (CBG). Questa proteina, tuttavia, possiede una bassa capacità di legame, che viene rapidamente superata quando la secrezione di cortisolo aumenta in condizioni di stress (14).
L’asse HPA è essenziale anche per il mantenimento dell’omeostasi in assenza di stress. CRH e AVP continuano ad essere secreti in maniera pulsatile e questo determina il rilascio del cortisolo secondo un ben definito ritmo circadiano (15), con picco di secrezione 30-40 minuti dopo il risveglio, successivo declino durante il resto della giornata e nadir che si osserva all’incirca all’inizio del sonno (16). Nei neonati di due mesi d’età è già possibile osservare una secrezione pulsatile di cortisolo (17), sebbene l’andamento adulto di secrezione venga raggiunto intorno al 3° anno di vita (18).
ASSE HPA E PATOLOGIA PSICHIATRICA
Possibile nesso causale
L’ampia capacità del cortisolo di agire su diversi sistemi neurotrasmettitoriali che regolano emozioni, motivazione e comportamento, ne suggerisce un ruolo nell’eziologia di numerose problematiche psichiatriche/comportamentali (5). Alcuni studi su animali sono a favore dell’ipotesi che l’esposizione a uno stress cronico possa condurre a cambiamenti persistenti nella regolazione dell’asse HPA, mediante mutamento dell’assetto neurotrasmettitoriale (19), e l’eventuale perdita del meccanismo di feedback negativo sembra possa favorire lo sviluppo di disturbi affettivi. Una disregolazione dell’asse HPA è infatti frequentemente osservata nei soggetti affetti da depressione maggiore (20), schizofrenia (21), disordini da stress post-traumatico (22) e dipendenza da alcool (23).
Altri dati derivanti da studi su animali e umani sembrano dimostrare che il cortisolo, sempre in condizione di stress ripetuto o duraturo, può esercitare un effetto neurotossico, con ricadute negative su funzioni esecutive, memoria, regolazione delle emozioni e performance cognitive (24,25).
Molto importante sembra la diversa risposta dei singoli individui allo stress, determinata dalla presenza di particolari varianti dei geni coinvolti nella funzione dell’asse HPA: CRH e suo recettore, GR, MR, FK506-binding protein (26).
Cortisolo e depressione
Molti studi hanno riportato un anomalo segnale del GR (22) nei pazienti con depressione maggiore, associato a cronica ipersecrezione di CRH (27). Tale situazione sposterebbe l’asse HPA verso un set-point più elevato, spiegando l’iperattività persistente osservata in alcuni pazienti affetti da depressione maggiore (28,29) o la mancata soppressione del cortisolo in risposta ai test farmacologici (30,31). Il risultante ipercortisolismo potrebbe inoltre modificare l’attività del sistema serotoninergico e dopaminergico, meccanismo che potrebbe spiegare i sintomi più severi o le manifestazioni psicotiche osservate in alcune forme di depressione maggiore (32,33).
Ma la relazione tra depressione e cortisolo è molto più complessa e appare dipendente dalla severità e stadio della malattia e anche dal tipo di metodo utilizzato nella sua valutazione (34). Rispetto alla gravità, sembra che nelle forme più severe di depressione (psicotica, melancolica, bipolare) i valori di cortisolo (basali, circadiani o dopo stimolo/soppressione) siano più elevati di quelli osservati nelle varianti mild (depressione atipica) (35-38). Elevati livelli di cortisolo vengono anche associati al deficit cognitivo osservato nei pazienti affetti da depressione psicotica (38). Inoltre, alcuni studi hanno mostrato diversità nella morfologia del SNC nei pazienti affetti da forme più gravi di depressione rispetto a quelli colpiti da sottotipi più mild: riduzione del volume dell’ippocampo e del lobo frontale, aumento della fessura silviana, riduzione della vascolarizzazione del lobo frontale (39-43).
Per quanto riguarda lo stadio della malattia, invece, la funzione dell’asse HPA non appare alterata nella depressione cronica, mentre i pazienti in fase di remissione sottoposti ad uno stress visivo mostrano un aumento dei valori di cortisolo maggiore rispetto ai controlli (44) e la persistenza di una mancata risposta alla soppressione con desametasone sembra predirne la ricaduta a sei mesi (45).
Prese tutte insieme, queste osservazioni suggeriscono che la disfunzione del cortisolo sia proporzionale alla corrente classificazione nosografica della depressione maggiore (37).
Cortisolo e schizofrenia
Gli studi sulla relazione tra asse HPA e schizofrenia hanno prodotto risultati non sempre concordanti. Alcuni sembrano indicare un’iperattività dell’asse, con valori di cortisolemia basale più elevati (46,47), mentre altri non hanno dimostrato alcuna differenza rispetto ai controlli (23). Più costantemente è riportata una ridotta risposta del cortisolo allo stress, indipendentemente da stadio di malattia, farmaci, o cronicità (23,48). Alcuni autori hanno inoltre suggerito che più elevati valori di cortisolemia basale sia associno a maggiore severità clinica, ridotta capacità di interazione sociale e delle performance cognitive (49,50).
EARLY LIFE PROGRAMMING E PATOLOGIA PSICHIATRICA
Nella relazione tra asse HPA e disturbi emotivi/comportamentali appare sempre più importante l’effetto dell’esposizione durante la vita intra-uterina e post-natale precoce a condizioni in grado di interagire da un lato con lo sviluppo e la maturazione del SNC e dall’altro con il sistema di risposta allo stress, fenomeno definito come “early-life programming” (51). Numerosi dati (52) indicano come uno stress materno durante la gravidanza o nei primi anni di vita post-natale aumenti il rischio per la prole di successive problematiche cognitive, comportamentali ed emotive, quali ansia, depressione, ADHD, disordini dello spettro autistico, disturbi della personalità e disturbi del comportamento (51,53,54). Alcuni lavori sembrano dimostrare che condizioni di stress in età infantile siano fortemente predittrici della presenza di un ridotto feed-back inibitorio dell’asse HPA (51) o comunque di un’alterazione della sua funzionalità. Uno studio su bambine e adolescenti tra 6 e 16 anni, vittime di abusi sessuali, ha dimostrato inizialmente valori di cortisolemia basale più elevati rispetto ai controlli, condizione che si invertiva in età adulta (55).
Il risultato dello stress pre- e post-natale sulla prole sembra dipendere da diversi fattori, quali il tipo di stress (56), il momento della vita in cui questo viene vissuto (57), il sesso del nascituro (58), oppure una specifica “vulnerabilità genetica” del bambino. Ad esempio, il rischio di schizofrenia sembra maggiormente associato a un intenso stress vissuto nel I trimestre di gravidanza (59), mentre la presenza di specifiche forma di COMT, l’enzima che metabolizza le catecolamine, pare associarsi alla diagnosi di ADHD in adolescenti figli di madri che hanno sofferto di ansia nel periodo della gravidanza (60). Un possibile meccanismo alla base di questa relazione potrebbe essere una modificazione epigenetica (metilazione del DNA, modificazioni post-traslazionali degli istoni, regolazione genetica attraverso microRNA) di quei geni associati con la regolazione dell’umore o la risposta allo stress (51), alterazioni che non solo possono potenzialmente modificare la risposta alle avversità del singolo individuo, ma possono essere anche trasmesse attraverso le generazioni (26,61,62). Nei piccoli di ratto sottratti alle cure materne è stata osservata l’esagerata metilazione di specifiche regioni del gene che codifica per GR (NR3C1) (61) e alcuni dati sembrano suggerire che tale alterazione possa modificare anche nell’uomo la funzione dell’asse HPA e aumentare il rischio di sviluppare disordini psichiatrici (63,64).
Numerosi studi hanno cercato di verificare come lo stress pre-natale possa interferire sull’anatomia del SNC (52). I risultati più interessanti sembrano dimostrare assottigliamento dello spessore della corteccia del lobo frontale e temporale (65,66), riduzione del volume della sostanza grigia (67) e del cervelletto, aumento del volume dell’amigdala (68) o ancora riduzione del volume dell’ippocampo (69) e della corteccia cingolata anteriore (70). Nei bambini l’assottigliamento della corteccia pre-frontale dell’emisfero di destra sembra associato a comportamenti esternalizzanti (72), mentre negli adolescenti l’assottigliamento della corteccia soprattutto frontale e temporale sembra associato a elevati sintomi depressivi (66). Tale alterazione dello sviluppo corticale è stata inoltre osservata nei pazienti affetti da depressione maggiore, indipendentemente dalla presenza di stress prenatale (72). L’amigdala è la struttura cerebrale coinvolta nella regolazione delle emozioni e un incremento del suo volume sembra collegarsi a disordini comportamentali (73).
Partendo dai lavori condotti sugli animali, i ricercatori hanno focalizzato la loro attenzione sull’asse HPA e sul ruolo del cortisolo quale possibile meccanismo sottostante alla relazione tra stress prenatale e l’insorgenza di successive problematiche comportamentali/psichiatriche. Durante la gravidanza il cortisolo materno aumenta di due-tre volte rispetto ai valori pre-gravidanza, con un picco di concentrazione al III trimestre, largamente dovuto alla produzione placentare di CRH. L’esposizione fetale al cortisolo materno è parzialmente limitata dalla azione dell’11ß-HSD2, ma di fatto i valori prenatali di cortisolo materno correlano con quelli fetali (74). Quindi, una madre che si trova a vivere uno stress prolungato e ripetuto potrebbe esporre il feto a livelli esagerati di cortisolo. I dati della letteratura sembrano indicare che nell’uomo l’esposizione a elevati livelli di cortisolo durante la gravidanza si associa a basso peso alla nascita, ridotta circonferenza cranica (75,76), ma anche a una più intensa risposta del cortisolo allo stress in epoca infantile e durante l’adolescenza, alterato sviluppo mentale e psico-motorio (77-79), minor QI, disordini da iperattività/deficit di attenzione, disordini affettivi, del comportamento e del controllo delle emozioni (80,81). I figli di madri che durante la gravidanza hanno consumato elevate quantità di liquirizia (che contiene un naturale inibitore della 11β-HSD2) e che quindi sono stati esposti ad elevate concentrazioni di cortisolo in utero, hanno un rischio tre volte maggiore di presentare sintomi di ADHD, minor QI e pubertà più precoce (82). Livelli elevati di cortisolo materno nelle fasi precoci della gravidanza sembrano associarsi a maggior volume dell’amigdala e più frequenti problemi affettivi nelle bambine (83), mentre è stata osservata un’associazione tra livelli di CRH materno durante la gravidanza e assottigliamento della corticale nella zona temporale frontale della prole (84).
Tutti questi lavori sembrano quindi suggerire che la relazione tra cortisolo e patologia psichiatrica abbia un’origine molto precoce, addirittura prenatale, e impongono un’importante riflessione sulle misure da adottare per prevenire o modificare il decorso di patologie così invalidanti.
BIBLIOGRAFIA
- Juster R-P, McEwen BS, Lupien SJ. Allostatic load biomarkers of chronic stress and impact on health and cognition. Neurosci Biobehav Rev 2010, 35: 2–16.
- Cone RD, Low MJ, Elmquist JK, Cameron JL. Williams Textbook of Endocrinology 2003.
- Groeneweg FL, Karst H, de Kloet ER, Joëls M. Mineralocorticoid and glucocorticoid receptors at the neuronal membrane, regulators of nongenomic corticosteroid signalling. Mol Cell Endocrinol 2012, 350: 299–309.
- Joels M, de Kloet ER. Mineralocorticoid and glucocorticoid receptors in the brain. Implications for ion permeability and transmitter systems. Prog Neurobiol 1994, 43: 1–36.
- Kamin HS, Kertes DA. Cortisol and DHEA in development and psychopathology. Horm Behav 2017, 89: 69–85.
- Carlberg C, Seuter S. Dynamics of nuclear receptor target gene regulation. Chromosoma 2010, 119: 479–84.
- Evanson NK, Herman JP, Sakai RR, Krause EG. Nongenomic actions of adrenal steroids in the central nervous system. J Neuroendocrinol 2010, 22: 846–61.
- Dallman MF, Pecoraro NC, la Fleur SE. Chronic stress and comfort foods: self-medication and abdominal obesity. Brain Behav Immun 2005, 19: 275–80.
- Joca SRL, Ferreira FR, Guimarães FS. Modulation of stress consequences by hippocampal monoaminergic, glutamatergic and nitrergic neurotransmitter systems. Stress 2009, 10: 227–49.
- Karst H, Joëls M. Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J Neurophysiol 2005, 94: 3479–86.
- Lee S, Kim H, Youm J, et al. Non-genomic effect of glucocorticoids on cardiovascular system. Pflugers Arch Eur J Physiol 2012, 464: 549–59.
- de Kloet E, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev 1998, 19: 269–301.
- Mora F, Segovia G, Del Arco A, et al. Stress, neurotransmitters, corticosterone and body–brain integration. Brain Res 2012, 1476: 71–85.
- Perogamvros I, Ray DW, Trainer PJ. Regulation of cortisol bioavailability — effects on hormone measurement and action. Nat Rev Endocrinol 2012, 8: 717–27.
- Chrousos G, Kino T, Charmandari E. Evaluation of the hypothalamic-pituitary-adrenal axis function in childhood and adolescence. Neuroimmunomodulation 2009, 16: 272–83.
- Smyth JM, Ockenfels MC, Gorin AA, et al. Individual differences in the diurnal cycle of cortisol. Psychoneuroendocrinology 1997, 22: 89–105.
- de Weerth C, van Hees Y, Buitelaar JK. Prenatal maternal cortisol levels and infant behavior during the first 5 months. Early Hum Dev 2003, 74: 139–51.
- Watamura SE, Donzella B, Kertes DA, Gunnar MR. Developmental changes in baseline cortisol activity in early childhood: relations with napping and effortful control. Dev Psychobiol 2004, 45: 125–33.
- Murgatroyd C, Spengler D. Epigenetics of early child development. Front Psychiatry 2011, 2: 16.
- Holsboer F. The corticosteroid receptor hypothesis of depression Neuropsychopharmacology 2000, 23: 477-501.
- Bradley AJ, Dinan TG. A systematic review of hypothalamic-pituitary-adrenal axis function in schizophrenia: implication for mortality. J Psychopharmacol 2010, 24 (4 suppl): 91-118.
- Morris MC, Compas BE, Garber J. Relations among posttraumatic stress disorders, comorbid major depression and HPA function: a systematic review and metanalysis. Clin Psychol Rev 2012, 32: 301-15.
- Stephens MAC, Wand G. Stress and the HPA axis: role of glucocorticoids in alcohol dependence. Alcohol Res 2012, 34: 468-83.
- de Quervain D. Glucocorticoid-induced inhibition of memory retrieval. Ann N Y Acad Sci 2006, 1071: 216-20.
- Sebastian V, Estil J, Chen D, et al. Acute physiological stress promotes clustering of synaptic markers and alters spine morphology in the hippocampus. PLoS One 2013, 8: e79077.
- Ferrer A, Labad J, Salvat-Pujol N, et al. Hypothalamic-pituitary-adrenal axis-related genes and cognition in major mood disorders: a systematic review. Progr Neuropsychopharmacol Biol Psychiatry 2020, 101: 109929.
- Horstmann S, Dose T, Lucae S, et al. Suppressive effect of mirtazapine on the HPA system in acutely depressed women seems to be transient and not related to antidepressant action. Psychoneuroendocrinology 2009, 34: 238–48.
- Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry 2003, 160: 1554–65.
- Young EA, Lopez JF, Murphy-Weinberg V, et al. Mineralocorticoid receptor function in major depression. Arch Gen Psychiatry 2003, 60: 24–8.
- Ising M, Künzel HE, Binder EB, et al. The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog Neuropsychopharmacol Biol Psychiatry 2005, 29: 1085–93.
- Dam H, Mellerup ET, Rafaelsen OJ. The dexamethasone suppression test in depression. J Affect Disord 1985, 8: 95–103.
- Leonard BE. The HPA and immune axes in stress: the involvement of the serotonergic system. Eur Psychiatry 2005, 20 (Suppl 3): S302–6.
- Schatzberg AF, Rothschild AJ, Langlais PJ, et al. A corticosteroid/dopamine hypothesis for psychotic depression and related states. J Psychiatr Res 1985, 19: 57–64.
- Nandam LS, Brazel M, Zhou M, Jahveri DJ. Cortisol and major depressive disorders – translating findings from humans to animal models and back. Front Psychiatry 2020, 10: 974.
- Posener JA, DeBattista C, Williams GH, et al. 24-Hour monitoring of cortisol and corticotrophin secretion in psychotic and nonpsychotic major depression. Arch Gen Psychiatry 2000, 57: 755–60.
- Sachar EJ, Hellman L, Roffwarg HP, et al. Disrupted 24-hour patterns of cortisol secretion in psychotic depressives. Arch Gen Psychiatry 1973, 28: 19–24.
- Keller J, Flores B, Gomez RG, et al. Cortisol circadian rhythm alterations in psychotic major depression. Biol Psychiatry 2006, 60: 275–81.
- Keller J, Gomez R, Williams G, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry 2017, 22: 527–36.
- Bijanki KR, Hodis B, Brumm MC, et al. Hippocampal and left subcallosal anterior cingulated atrophy in psychotic depression. PLoS One 2014, 9: e110770.
- Hickie I, Naismith S, Ward PB, et al. Reduced hippocampal volumes and memory loss in patients with early- and late-onset depression. Br J Psychiatry 2005, 186: 197–202.
- Simpson S, Baldwin RC, Jackson A, Burns A. The differentiation of DSM-III-R psychotic depression in later life from nonpsychotic depression: comparisons of brain changes measured by multispectral analysis of magnetic resonance brain images, neuropsychological findings, and clinical features. Biol Psychiatry 1999, 45: 193–204.
- Pujol J, Cardoner N, Benlloch L, et al. CSF spaces of the sylvian fissure region in severe melancholic depression. Neuroimage 2002, 15: 103–6.
- Fountoulakis KN, Iacovides A, Gerasimou G, et al. The relationship of regional cerebral blood flow with subtypes of major depression. Prog Neuropsychopharmacol Biol Psychiatry 2004, 28: 537–46.
- Holsen LM, Lancaster K, Klibanski A, et al. HPA-axis hormone modulation of stress response circuitry activity in women with remitted major depression. Neuroscience 2013, 250: 733–42.
- Zobel AW, Nickel T, Sonntag A, et al. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression: a prospective study. J Psychiatr Res 2001, 35: 83–94.
- Breier A, Buchanan RW. The effects of metabolic stress on plasma progesterone in healthy volunteers and schizophrenic patients. Life Sci 1992, 51: 1527-34.
- Walker EF, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol 2008, 4: 189-216.
- Zorn JV. Schur RR, Boks MP, et al. Cortisol stress reactivity across psychiatric disorders: a systematic review and metanalysis. Psychoneuroendocrinology 2017, 77: 25-36.
- Walder DJ, Walker EF, Lewine RJ. Cognitive functioning, cortisol release, and symptom severity in patients with schizophrenia. Biolog Psychiatry 2000, 48: 1121-32.
- Hempel RJ, Tulen JHM, van Beveren NJM, et al. Diurnal cortisol patterns of young male patients with schizophrenia. Psychiatry Clin Neurosci 2010, 64: 548-54.
- Jawahar MC, Murgatroyd C, Harrison EL, Baune BT. Epigenetic alterations following early postnatal stress: a review on novel aetiological mechanism of common psychiatric disorders. Clin Epigenetics 2015, 7: 122.
- Lautarescu A, Craig MC, Glover V. Prenatal stress: effects on fetal and child brain development. Int Rev Neurobiol 2020, 150: 17-40.
- Green JG, McLaughlin KA, Berglund PA, et al. Childhood adversities and adult psychiatric disorders in the national comorbidity survey replication I. Associations with first onset of DSM-IV disorders. Arch Gen Psychiatry 2010, 67: 113-23.
- Varese F, Smeets F, Drukker M, et al. Childhood adversities increase the risk of psychosis: a meta-analysis of patient-control, prospective- and cross-sectional cohort studies. Schizophr Bull 2012, 38: 661–71.
- Badanes LS, Watamura SE, Hankin BL. Hypocortisolism as a potential marker of allostatic load in children: associations with family risk and internalizing disorders. Dev Psychopathol 2011, 23: 881–96.
- Gershon A, Sudheimer K, Tirouvanziam R, et al. The long-term impact of early adversity on late-life psychiatric disorders. Curr Psychiatry Rep 2013, 15: 352.
- Evans GW, Schamberg MA. Childhood poverty, chronic stress, and adult working memory. Proc Natl Acad Sci 2009, 106: 6545-9.
- Glover V, Hill J. Sex differences in the programming effects of prenatal stress on psychopathology and stress responses: an evolutionary perspective. Physiol Behav 2012, 106: 736–40.
- Guo C, He P, Song X, Zheng X. Long-term effects of prenatal exposure to earthquake on adult schizophrenia. Br J Psychiatry 2019, 215: 730-5.
- O’Donnell KJ, Glover V, Lahti J, et al. Maternal prenatal anxiety and child COMT genotype predict working memory and symptoms of ADHD. PLoS One 2017, 12: e0177506.
- Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci 2004, 7: 847–54.
- McGowan P, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009, 12: 342–8.
- Palma-Gudiel H, Cordova-Palomera A, Leza JC, Fananas L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav Rev 2015, 55: 520–35.
- Palma-Gudiel H, Cordova-Palomera A, Tornador C, et al. Increased methylation at an unexplored glucocorticoid responsive element within exon 1D of NR3C1 gene is related to anxious-depressive disorders and decreased hippocampal connectivity. Eur Neuropsychopharmacol 2018, 28: 579–88.
- Lebel C, Walton M, Letourneau N, et al. Prepartum and postpartum maternal depressive symptoms are related to children’s brain structure in preschool. Biol Psychiatry 2016, 80: 859–68.
- Davis EP, Hankin BL, Glynn LM, et al. Prenatal maternal stress, child cortical thickness, and adolescent depressive symptoms. Child Dev 2020, 91: e432-50.
- Favaro A, Tenconi E, Degortes D, et al. Neural correlates of prenatal stress in young women. Psychol Med 2015, 45: 2533–43.
- Acosta H, Tuulari JJ, Scheinin NM, et al. Maternal pregnancy-related anxiety is associated with sexually dimorphic alterations in amygdala volume in four-year-old children. Front Behav Neurosci 2019, 13: 175.
- Qiu A, Rifkin-Graboi A, Chen H, et al. Maternal anxiety and infants’ hippocampal development: timing matters. Transl Psychiatry 2013, 3: e306.
- Mareckova K, Klasnja A, Bencurova P, et al. Prenatal stress, mood, and gray matter volume in young adulthood. Cereb Cortex 2019, 29: 1244–50.
- Sandman CA, Buss C, Head K, Poggi Davis E. Fetal exposure to maternal depressive symptoms is associated with cortical thickness in late childhood. Biol Psychiatry 2015, 77: 324–34.
- Suh JS, Schneider MA, Minuzzi L, et al. Cortical thickness in major depressive disorder: a systematic review and meta-analysis. Progr Neuropsychopharmacol Biol Psychiatry 2019, 88: 287–302.
- Jones SL, Dufoix R, Laplante DP, et al. Larger amygdala volume mediates the association between prenatal maternal stress and higher levels of externalizing behaviors: sex specific effects in project ice storm. Front Hum Neurosci 2019, 13: 144.
- Field T, Diego M, Hernadez-Reif M, et al. Prenatal maternal biochemistry predicts neonatal biochemistry. Int J Neurosci 2004, 114: 933-45.
- Baibazarova E, van de Beek C, Cohen-Kettenis PT, et al. Influence of prenatal maternal stress, maternal plasma cortisol and cortisol in amniotic fluid on birth outcomes and child temperament at 3 months. Psychoneuroendocrinology 2013, 38: 907-15.
- Bolten M, Wurmser H, Buske-Kirschbaum A, et al. Cortisol level in pregnancy as a psychobiological predictor for birth weight. Arch Women Ment Health 2011, 14: 33-41.
- O’Connor T, Bergaman K, Sarkar P, Glover V. Prenatal cortisol exposure predicts infant cortisol response to acute stress. Dev Psychobiol 2013, 55: 145-55.
- Werner E, Zhao Y, Evans L, et al. Higher maternal prenatal cortisol and younger age predict greater infancy reactivity to novelty at 4 months: an observation-based study. Dev Psychobiol 2013, 55: 707-18.
- Zijlmans MC, Riksen-Walraven JM, de Weerth C. Association between maternal prenatal cortisol concentration and child outcome: a systematic review. Neurosci Biobehav Rev 2015, 53: 1-24.
- Rice F, Harold GT, Boivin J, et al. The links between prenatal stress and offspring development and psychopathology: disentangling environmental and inherited influences. Psychol Med 2010, 40: 335-45.
- Barker ED, Maughan B. Differentiating early-onset versus childhood-limited conduct problem youth. Am J Psychiatry 2009, 166: 900-8.
- Raikkonen K, Martikainen S, Pesonen AK, et al. Maternal licorice consumption during pregnancy and pubertal, cognitive, and psychiatric outcomes in children. Am J Epidemiol 2017, 185: 317–28.
- Graham AM, Rasmussen JM, Entringer S, et al. Maternal cortisol concentrations during pregnancy and sex-specific associations with neonatal amygdala connectivity and emerging internalizing behaviors. Biol Psychiatry 2019, 85: 172–81.
- Sandman CA, Curran MM, Poggi-Davis E, et al. Cortical thinning and neuropsychiatric outcomes in children exposed to prenatal adversity: a role for placental CRH? Am J Psychiatry 2018, 175: 471–9.
ESTROGENI E PSICHIATRIA
EFFETTI DEGLI ESTROGENI SUL SNC
Gli ormoni gonadici non indirizzano soltanto lo sviluppo somatico ma anche quello cerebrale, creando le basi per un dimorfismo sessuale che inizia già in epoca gestazionale e si concretizza ancora più fortemente in epoca puberale. Gli estrogeni, e in particolare il 17ß-estradiolo (E2), sembrano garantire il mantenimento di una normale funzione cerebrale durante tutta la durata della vita attraverso diversi meccanismi: promuovono la mielinizzazione neuronale, incrementano plasticità e densità delle sinapsi, facilitano la connessione neuronale, espletano un ruolo anti-ossidante, inibiscono la morte neuronale, mediano l’espressione del BDNF e influenzano positivamente l’attività mitocondriale neuronale (1,2).
Gli estrogeni possono certamente avere un’influenza sull’umore e sulla regolazione cognitiva, grazie all’ampia diffusione dei loro recettori nel cervello e alla capacità di modulare sintesi e attività di molteplici neurotrasmettitori. In particolare, per la regolazione dell’umore appare importante la presenza di recettori estrogenici in regioni quali la corteccia pre-frontale e l’ippocampo (3,4).
Gli studi condotti su animali evidenziano che la somministrazione di E2 è in grado di determinare un incremento della sintesi e disponibilità della serotonina, attraverso la limitazione dell'attività della monoamino-ossidasi (MAO) e l’incremento delle diverse forme di triptofano-idrossilasi, enzimi coinvolti su fronti opposti nel metabolismo di questo neurotrasmettitore (5-7). Un ulteriore effetto anti-depressivo potrebbe essere esercitato dagli estrogeni attraverso la loro modulazione positiva del BDNF (8).
SCHIZOFRENIA
La schizofrenia rappresenta uno dei più comuni disturbi psichiatrici che colpiscono la nostra società, interessando circa 7 individui su 1000 (9).
Il suo esordio avviene generalmente in età adolescenziale, persiste per tutta la vita ed è caratterizzata da sintomi positivi (delusione, allucinazioni), negativi (comportamenti inappropriati, isolamento sociale, apatia, disorganizzazione) e deficit cognitivi, generalmente a largo spettro, che possono interessare memoria, attenzione e funzioni esecutive (9-11).
I maschi presentano un maggiore rischio di malattia prima dei 40 anni (RR 1.5) (12,13), mentre il rischio per le donne è più alto nel periodo peri-menopausale (14).
Sebbene i risultati degli studi non siano sempre omogenei, le donne prima della menopausa sembrano presentare un decorso meno grave rispetto agli uomini, con minor frequenza di disturbi cognitivi (9), minor numero di ricoveri (15) e minor dosaggio di neurolettici (16). Esistono anche altre differenze tra i sessi: gli uomini affetti sembrano mostrare una riduzione del volume del lobo temporale sinistro e del lobo parietale inferiore rispetto a soggetti sani dello stesso sesso, mentre tale differenza non è stata osservata nelle donne (17,18). Il migliore decorso di malattia osservato nelle donne potrebbe essere spiegato da un lato dalla migliore compliance e dal minore abuso di sostanze che spesso le pazienti mostrano rispetto ai maschi, ma anche da un possibile ruolo protettivo esercitato dagli estrogeni. Gli estrogeni giocano un ruolo importante nella modulazione di diversi sistemi neurotrasmettitoriali (dopaminergico, GABAergico, serotoninergico, noradrenergico e colinergico), tutti coinvolti nello sviluppo e nell’espressione della sintomatologia schizofrenica (2,19-21). Questi dati, associati alle osservazioni precedenti, hanno suggerito per gli estrogeni, soprattutto E2, sia un ruolo di protezione rispetto all’insorgenza della schizofrenia sia di modulazione positiva di alcuni sintomi psicotici. Il brusco declino di questi ormoni, che si verifica con la menopausa, potrebbe essere responsabile del secondo picco di incidenza della patologia che si osserva in questa fase della vita (22). Peraltro già nelle donne sane il periodo peri- e post-menopausale sembra associato a una riduzione della memoria verbale e della fluenza fonemica, oltre che a un aumentato rischio di depressione (23). Ad ulteriore sostegno di tale ipotesi, alcuni ricercatori hanno evidenziato nelle donne schizofreniche in età fertile livelli di estrogeni più bassi di quelli rilevati nelle donne sane, così come un esordio o riacutizzazione di malattia in quelle fasi del ciclo mestruale nelle quali questi ormoni sono fisiologicamente ridotti o nel post-gravidanza (24). Le donne schizofreniche con ciclo regolare sembrano mostrare funzioni cognitive superiori rispetto a quelle affette dalla stessa patologia ma con cicli irregolari (25) e nelle stesse pazienti l’età del menarca sembra inversamente correlata con l’età di insorgenza della patologia (16).
Azione terapeutica degli estrogeni e dei SERM
Diversi studi clinici di breve durata, condotti in donne in età fertile o in menopausa, hanno dimostrato come il trattamento con estrogeni (E2 per via orale o trans-dermica), da soli o in aggiunta alla terapia anti-psicotica, sia in grado di migliorare tutti i diversi sintomi della schizofrenia (positivi, negativi, cognitivi) (26-30). Pochi studi hanno invece valutato l’effetto della terapia combinata (estrogeni + progestinici) con risultati non univoci (31,32), sebbene in queste pazienti appaia evidente anche una disregolazione dei livelli di progesterone (33).
Se da un lato, tuttavia, la prescrizione di estrogeni per un breve periodo si è dimostrata efficace e sicura, dall’altro il trattamento a lungo termine rimane controverso. Nelle donne in menopausa la terapia con soli estrogeni aumenta il rischio di neoplasia mammaria ed endometriale (34,35), sebbene offra una protezione contro la malattia cardio-vascolare (36) e appaia ridurre il declino cognitivo (37).
Poiché le donne con schizofrenia richiedono un trattamento cronico, l’interesse dei ricercatori si è rivolto quindi verso i SERM (selective estrogen receptor modulators), che esplicano un effetto estrogenico ed anti-estrogenico a secondo del tessuto con cui interagiscono. Tra questi, il più studiato è il raloxifene, con un effetto anti-estrogenico su mammella e utero (38) e azione agonista sui recettori serotoninergici nei gangli basali, corteccia frontale e striato, tutti siti coinvolti nell’eziologia/sintomatologia della schizofrenia (39). Come per gli estrogeni, gli studi al momento disponibili sono di breve durata (8-24 settimane) e sembrano indicare efficacia e sicurezza del raloxifene (60-120 mg/die), da solo o in associazione alla terapia anti-psicotica, nel trattamento delle donne in menopausa affette da schizofrenia (40,41). Tuttavia, i risultati degli studi non sono sempre concordi, probabilmente perché influenzati da variabili difficilmente confrontabili, che possono modulare la risposta al raloxifene, come età di insorgenza, durata e gravità della patologia, tipo di anti-psicotico utilizzato, dosaggio del SERM, profilo genetico della paziente (42). Al momento il raloxifene non è raccomandato per il trattamento della schizofrenia, se non in studi sperimentali, sebbene gli studi appaiano promettenti. Gli anti-psicotici, quindi, rimangono l’unico trattamento possibile per le donne in menopausa, sebbene tali farmaci si mostrino meno efficaci in questa fase della vita e più frequentemente accompagnati da effetti collaterali di tipo metabolico, cardio-vascolare e neurologico (43).
DEPRESSIONE
La depressione rappresenta una condizione disabilitante, che incide profondamente sulla vita personale, affettiva e sociale di un individuo e che interessa circa 300 milioni di persone nel mondo. I sintomi, che insorgono già in adolescenza, possono essere di tipo psicologico (sentirsi giù, tristi, senza speranza, privi di motivazione o interessi, irritabili, colpevoli, privi di stima per se stessi, avere fantasie suicidarie), fisico (cambiamenti della qualità e della durata del sonno, modificazione del peso e dell’appetito, perdita di energia, dolori inspiegabili, perdita di interesse per la sessualità) e sociale (ridotta capacità lavorativa, evitamento della socialità, difficoltà nella vita familiare) (44).
La depressione rappresenta una delle cause più importanti di disabilità per le donne, che presentano un rischio quasi doppio di ammalarsi rispetto agli uomini, soprattutto durante la vita riproduttiva (45). Tale predisposizione appare influenzata da fattori sociali, psicologici e genetici, così come da fattori biologici. Infatti, alcune donne sembrano sperimentare una maggiore vulnerabilità alla depressione in alcune fasi della loro vita (finestre di vulnerabilità), quali il periodo luteale, il post-partum o la fase di transizione verso la menopausa (46). Questi periodi “finestra” sono caratterizzati da modificazioni dell’ambiente ormonale al quale queste donne sembrano particolarmente sensibili. L’importanza e l’attenzione che viene rivolta a queste finestre di vulnerabilità è testimoniata anche dal riconoscimento del “disordine disforico premestruale” e dalla sua inclusione nel DSM-V (47).
L’esistenza di una depressione associata alla menopausa appare invece ancora un punto controverso. Diversi studi (48-50), sia trasversali che longitudinali, sembrano dimostrare un incremento del rischio di depressione nel periodo di transizione verso la menopausa (da due a quattro volte rispetto alla pre-menopausa) e identificano una serie di fattori predisponenti, quali la presenza di sintomi vasomotori, l’abitudine tabagica, lo stato di salute, i disturbi del sonno, precedenti episodi di depressione maggiore. Per quanto riguarda la relazione con l’assetto ormonale, i dati non sono sempre concordi. Il Penn Ovarian Ageing study (49) sembra mettere in relazione le fluttuazioni dei valori di FSH ed estradiolo che si osservano in peri-menopausa con un peggioramento dello stato dell’umore, a sostegno dell’ipotesi che l’impatto sul rischio di malattia sia esercitato maggiormente dalle variazioni piuttosto che dal valore assoluto degli ormoni sessuali. L’aumento del rischio di depressione in menopausa è stato associato a un polimorfismo per il recettore per gli estrogeni (51-53), sebbene gli studi di popolazione che hanno valutato il ruole di tale variazione allelica abbiano prodotto conclusioni non univoche.
Terapia estrogenica per la depressione
Sebbene ci siano pochi studi randomizzati che hanno esaminato l’efficacia della somministrazione di estradiolo in donne con esordio o ripresa di depressione in fase peri-menopausale (talvolta con risultati difficilmente confrontabili), i dati della letteratura sembrano promettenti, indicando un’azione anti-depressiva di questo ormone somministrato per via orale o trans-dermica, anche in associazione a un progestinico (54-57). L’efficacia sembra però limitata nelle donne ormai in menopausa da molti anni (58). Altri studi suggeriscono che l’estradiolo possa aumentare la risposta clinica agli anti-depressivi (SSRI) o in taluni casi avere una efficacia sovrapponibile nella riduzione dei sintomi depressivi, con un’azione apparentemente svincolata dal controllo delle vampate e dal miglioramento del sonno (59,60). Tuttavia, anche in questo caso si tratta di studi di breve durata, condotti in genere su campioni non troppo grandi di donne e di età talvolta diverse, il che rende impegnativo tradurre questi risultati in indicazioni operative/terapeutiche concrete, sebbene possano ampliare il nostro armamentario terapeutico e condurci a un trattamento sempre più personalizzato delle nostre pazienti in menopausa.
BIBLIOGRAFIA
- Riecher-Rossler A. Oestrogen, prolactin, hypotalamic-pituitary-gonadal axis and schizophrenic psychoses. Lancet Psychiatry 2017, 4: 63-72.
- Shimamoto A, Rappenau V. Sex dependent mental illnesses and mitochondria. Schizophr Res 2017, 187: 38-46.
- Deecher D, Andree TH, Sloan D, Schechter LE. From menarche to menopause: exploring the underlying biology of depression in women experiencing hormonal changes. Psychoneuroendocrinology 2008, 33: 3-17.
- Genazzani AR, Bernardi F, Pluchino N, et al. Endocrinology of menopausal transition and its brain implication. CNS Spectr 2005, 10: 449-57.
- Gundlah C, Lu NZ, Bethea CL. Ovarian steroid regulation of monoamine oxidase A and B mRNA in the macaque dorsal raphe and hypothalamic nuclei. Psychopharmacology (Berl) 2002, 160: 271-82.
- Bethea CL, Mirkes SJ, Su A, Michelson D. Effects of oral estrogen, raloxifene and arzoxifene on gene expression in serotonin neurons of macaque. Psychoneuroendocrinology 2002, 27: 431-5.
- Hiroi R, McDevitt RA, Neumaier JF. Estrogen selectively increases tryptophan hydroxylase-2 mRNA expression in distinct subregions of rat midbrain raphe nucleus: association beetween gene expression and anxiety behavior in the open field. Biol Psychiatry 2006, 60: 288-95.
- Srivastava DP, Woolfrey KM, Evans PD. Mechanisms underlying the interactions between rapid estrogenic and BDNF control of synaptic connectivity. Neuroscience 2013, 239: 17-33.
- Searles S, et al. The role of estradiol in schizophrenia diagnosis and symptoms in postmenopausal women. Schizophr Res 2018, 196: 35–8.
- Buoli M, et al. Prominent clinical dimension, duration of illness and treatment response in schizophrenia: a naturalistic study. Psychiatry Investig 2012, 9: 354–60.
- van Os J, Kapur S. Schizophrenia. Lancet 2009, 374: 635–45.
- Aleman A, Kahn RS, Selten JP. Sex differences in the risk of schizophrenia. Evidence from meta-analysis. Arch Gen Psychiatry 2003, 60: 565–71.
- McGrath J, et al. A systematic review of the incidence of schizophrenia: the distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med 2004, 2: 13.
- Hafner H. Gender differences in schizophrenia. Psychoneuroendocrinology 2003, 28: 17–54.
- Ochoa S, et al. Gender differences in schizophrenia and first-episode psychosis: a comprehensive literature review. Schizophr Res Treatment 2012, 2012: 916198.
- Gonzalez-Rodriguez A, et al. Antipsychotic response worsens with postmenopausal duration in women with schizophrenia. J Clin Psychopharmacol 2016, 36: 580–7.
- Bryant NL, et al. Gender differences in temporal lobe structures of patients with schizophrenia: a volumetric MRI study. Am J Psychiatry 1999, 156: 603–9.
- Frederikse M, et al. Sex differences in inferior parietal lobule volume in schizophrenia. Am J Psychiatry 2000, 157: 422–7.
- Sanchez MG, Morissette M, Di PT. Oestradiol modulation of serotonin reuptake transporter and serotonin metabolism in the brain of monkeys. J Neuroendocrinol 2013, 25: 560-9.
- Meitzen J, Mermelstein PG. Estrogen receptors stimulate brain specific metabotropic glutamate receptors to rapidly initiate signal transduction pathways. J Chem Neuroanat 2011, 42: 236-41.
- Barth C, Villringer A, Sacher J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front Neurosci 2015, 9: 37.
- Riecher-Rossler A, Butler S, Kulkarni J. Sex and gender differences in schizophrenic psychoses – a critical review. Arch Womens Ment Health 2018, 21: 627-48.
- Weber MT, et al. Cognition and mood in perimenopause: a systematic review and meta-analysis. J Steroid Biochem Mol Biol 2014, 142: 90–8.
- Riecher-Rossler A, Kulkarni J. Estrogens and gonadal function in schizophrenia and related psychoses. Curr Top Behav Neurosci 2011, 8: 155-71.
- Gleeson PC, et al. Menstrual cycle characteristics in women with persistent schizophrenia. Aust N Z J Psychiatry 2016, 50: 481–7.
- Kulkarni J, De Castella A, Smith D, et al. A clinical trial of the effects of estrogen in acutely psychotic women. Schizophr Res 1996, 20: 247–52.
- Kulkarni J, Riedel A, De Castella AR, et al. Estrogen - a potential treatment for schizophrenia. Schizophr Res 2001, 48: 137–44.
- Kulkarni J, De Castella A, Fitzgerald PB, et al. Estrogen in severe mental illness: a potential new treatment approach. Arch Gen Psychiatry 2008, 65: 955–60.
- Kulkarni J, Riedel A, De Castella AR, et al. A clinical trial of adjunctive oestrogen treatment in women with schizophrenia. Arch Womens Ment Health 2002, 5: 99–104.
- Akhondzadeh S, Nejatisafa AA, Amini H, et al. Adjunctive estrogen treatment in women with chronic schizophrenia: a double-blind, randomized, and placebo-controlled trial. Prog Neuropsychopharmacol Biol Psychiatry 2003, 27: 1007–12.
- Baxtera MG, et al. Multiple clinically relevant hormone therapy regimens fail to improve cognitive function in aged ovariectomized rhesus monkeys. Neurobiol Aging 2013, 34: 1882-90.
- Ko YH, Joe SH, Cho W, et al. Effect of hormone replacement therapy on cognitive function in women with chronic schizophrenia. Int J Psichiatry Clin Pract 2006, 10: 97-104.
- Sun J, Walker AJ, Dean B, et al. Progesterone: the neglected hormone in schizophrenia? A focus on progesterone-dopamine interactions. Psiconeuroendocrinology 2016, 74: 126-40.
- Santen RJ, Kagan R, Altomare CJ, et al. Current and evolving approaches to individualizing estrogen receptor-based therapy for menopausal women. J Clin Endocrinol Metab 2014, 99: 733–47.
- Hickey M, McNamara HC, Mishra GD. Menopausal estrogen therapy and breast cancer mortality. JAMA 2018, 319: 193.
- Arnson Y, Rozanski A, Gransar H, et al. Hormone replacement therapy is associated with less coronary atherosclerosis and lower mortality. J Am Coll Cardiol 2017, 69 Suppl DOI: 10.1016/ S0735-1097(17)34797-6.
- McGregor C, Riordan A, Thornton J. Estrogens and the cognitive symptoms of schizophrenia: possible neuroprotective mechanisms. Front Neuroendocrinol 2017, 47: 19–33.
- Moen MD, Keating GM. Raloxifene. A review of its use in the prevention of invasive breast cancer. Drugs 2008, 68: 2059-83.
- Littleton-Kearney MT, Ostrowski NL, Cox DA, et al. Selective estrogen receptor modulators: tissue action and potenzial for CNS protection. CNS Drug Rev 2002, 8: 309-30.
- De Boer J, Prikken M, Lei WU, et al.The effect of raloxifene augmentation in men and women with a schizophrenia spectrum disorder: a systematic review and meta-analysis. NPJ Schizophr 2018, 4: 1.
- Wang Q, Dong X, Wang Y, Li X. Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a meta-analysis of randomized controlled trials. Arch Womens Ment Health 2017, 21: 31-41.
- Labad J, Martorell L,Huerta-Ramos E, et al. Pharmacogenetic study of the effects of raloxifene on negative symptoms of postmenopausal women with schizophrenia: a double-blind, randomized, placebo controlled trial. Eur Neuropsychopharmacol 2016, 26: 1683–9.
- Seeman MV. Secondary effects of antipsychotics: women at greater risk than men. Schizophr Bull 2009, 35: 937–48.
- Sassarini J. Depression in midlife women. Maturitas 2016, 94: 149-54.
- Kessler RC, et al. Sex and depression in the National Comorbidity Survey. I: lifetime prevalence chronicity and recurrence. J Affect Disord 1993, 29: 85-96.
- Noble RE. Depression in women. Metabolism 2005, 54 (5 suppl 1): 49-52.
- Soares CN. Depression and menopause: current knowledege and clinical recommendations for a critical windows. Psychiatr Clin N Am 2017, 40: 239-54.
- Cohen LS, et al. Risk for new onset of depression during the menopausal transition. The Harvard study of moods and cycle. Arch Gen Psychiatr 2006, 63: 385-90.
- Freeman EW, Sammel MD, Lin H, Nelson DB. Association of hormones and menopausal status with depressed mood in women with no history of depression. Arch Gen Psychiatr 2006, 63: 375-82.
- Bromberger JT, Kravitz HM. Mood and menopause: findings from the study of Women’s Health Across the Nation (SWAN) over 10 years. Obstet Gynecol Clin North Am 2011, 38: 609-25.
- Kim JJ, et al. Association between estrogen receptor gene polymorphism and depression in post-menopausal women. Psychiatr Invest 2010, 7: 224-7.
- Keyes K, et al. The role of allelic variation in estrogen receptor genes and major depression in the Nurses Health Study. Soc Psychiatry Psychiatr Epidemiology 2015, 12: 1893-904.
- Ryan J, Ancelin ML. Polymorphism of estrogen receptor and risk of depression. Drugs 2012, 72: 1725-38.
- Cohen LS, Soares CN, Poitras JR, et al. Short term use of estradiol for depression in perimenopausal and postmenopausal women: a preliminary report. Am J Psychiatry 2003, 160: 1519-22.
- Ragson NL Altshulder LL, Faibanks L. Estrogen replacement therapy for depression. Am J Psychiatry 2001, 158: 1738.
- Soares CN, Almeida OP, Joffe H, Cohen LS. Efficacy of estradiol for treatment of depressive disorders in perimenopausal women: a double blind, randomized placebo-controlled trial. Arch Gen Psychiatry 2001, 58: 529-34.
- Rudolph I, Palombo-Kinne E, Kirsch B, et al. Influence of a continuos combined HRT (2 mg estradiol valerate and 2 mg dienogest) on post menopausal depression. Climateric 2004, 7: 301-11.
- Morrison MF, Kallan MJ, Ten Have T, et al. Lack of efficacy of estradiol for depression in postmenopausal women: a randomized controlled trial. Biol Psychiatry 2004, 55: 406-12.
- Amsterdam J, Garcia-Espana F, Fawcett J, et al. Fluoxetine efficacy in menopausal women with and without estrogen replacement. J Affect Disord 1999, 55: 11-7.
- Joffe H, Fagioli-Petrillo L, Koukopoulos A, et al. Increased estradiol and improved sleep, but not hot flashes, predict enhanced mood during the menopausal transition. J Clin Endocrinol Metab 2011, 96: E1044-54.
ANDROGENI E PSICHIATRIA
EFFETTI DEL TESTOSTERONE SUL SNC
La prima funzione del testosterone si manifesta durante la vita prenatale, quando è indispensabile per indurre la differenziazione sessuale e lo sviluppo del pene, dei testicoli e dell’apparato genitale maschile (1). Il testosterone svolge anche un ruolo importante nella differenziazione sessuale del SNC, dove interviene in senso dimorfico proprio sulla modulazione della funzione riproduttiva, sessuale, cognitiva e psicologica (2). La sua azione viene esplicata sia direttamente, attraverso recettori specifici (recettori androgenici, AR), sia indirettamente attraverso i recettori estrogenici, dopo la sua aromatizzazione in estrogeni da parte dell’aromatasi cerebrale. Inoltre, sempre nel tessuto cerebrale il testosterone può agire dopo trasformazione in DHT, che si comporta come ligando per gli AR (3).
Gli studi sugli animali e sull’uomo hanno dimostrato che l’espressione degli AR è specifica per le diverse regioni cerebrali (4) e dipendente dal genere, quindi con differente attivazione in funzione del sesso (5). Nel SNC dei mammiferi gli AR sono localizzati in aree critiche per le funzioni cognitive e l’umore, come la corteccia frontale, l’ippocampo, l’amigdala e l’ipotalamo (6,7), ma sono stati osservati anche in regioni cerebrali coinvolte nell’eziologia delle psicosi, come i neuroni dopaminergici del mesencefalo (8). Il testosterone sembra inoltre in grado di agire direttamente, e sempre in modo dimorfico, sulla morfologia delle strutture cerebrali corticali (9), e insieme al DHEA sembra regolare neurogenesi e apoptosi nelle strutture limbiche, soprattutto amigdala e ippocampo (10).
TESTOSTERONE E PSICHIATRIA
È stato ormai dimostrato che esistono differenze di genere per quanto riguarda esordio, incidenza, sintomi e decorso di malattie quali schizofrenia e disturbi dell’umore (depressione e disturbi bipolari) (11,12): mentre per gli estrogeni è stato proposto un effetto neuro-protettivo (13), appare meno chiaro il ruolo del testosterone in termini di prevenzione o induzione della patologia psichiatrica.
Diversi dati sembrano indicare che il testosterone possa mediare o favorire la sintomatologia neuropsichiatrica. Ad esempio, gli studi condotti su soggetti che abusano di androgeni sintetici a scopo anabolizzante, indicano che elevati livelli di androgeni possono favorire l’insorgenza di psicosi, aggressività, ansia (14-16), oltre ad esercitare un possibile effetto neurotossico con degenerazione neuronale e apoptosi (17).
Per quanto riguarda la schizofrenia, alcuni autori hanno riportato nei pazienti affetti un'associazione tra bassi livelli di testosterone e sintomi negativi più severi, peggiore funzione cognitiva ed alterata elaborazione delle emozioni (18-20).
Molto più numerosi sono i dati che tentano di stabilire un nesso di tipo causale tra testosterone e disturbi del tono dell’umore. Nei giovani pazienti con ipogonadismo congenito viene riportata elevata incidenza di disfunzioni sessuali, ansia e depressione, sulle quali la terapia sostitutiva con testosterone sembra avere un impatto decisamente positivo (21). Anche la terapia di deprivazione androgenica (ADT), utilizzata nel trattamento dei pazienti con carcinoma della prostata, sembra indurre la comparsa o il peggioramento di disturbi depressivi (22,23) e un recente studio australiano ha osservato un rischio di sviluppare depressione nei pazienti in ADT 10 volte maggiore rispetto ai controlli (24).
Negli uomini il declino dei valori di testosterone legato all’età è associato a sintomi di tipo depressivo (25), che la terapia sostitutiva sembra migliorare (26), come confermato anche da una recente meta-analisi (27).
Da queste pubblicazioni non possiamo però estrapolare la conclusione che la terapia con testosterone sia in grado di indurre la remissione di un disturbo depressivo maggiore o che possa facilitare la risposta alla terapia anti-depressiva. Infatti, la maggior parte di questi dati si riferisce a pazienti “endocrinologici”, che per vari motivi hanno ridotti livelli di testosterone, ma non una diagnosi psichiatrica di depressione, e nei quali l’effetto sull’umore della terapia sostitutiva è generalmente valutato attraverso questionari auto-somministrati (28). Anche nei “Testosterone Trial” (29), un gruppo di sette studi coordinati controllati con placebo, condotti su uomini anziani con ridotti livelli di testosterone, nel braccio in terapia sostitutiva si è osservato un piccolo ma statisticamente significativo miglioramento dell’umore e dei sintomi depressivi. Tuttavia, i partecipanti al “Testosterone Trial” non sono stati selezionati sulla base di una diagnosi di disturbo depressivo. I risultati possono quindi solo suggerire che una non meglio definita condizione depressiva associata all’ipogonadismo, non necessariamente sovrapponibile alla depressione maggiore, può essere migliorata dalla terapia con testosterone. Per spiegare questi risultati è stata ipotizzata l’esistenza di un disturbo distimico late-onset, non associato a coesistente depressione maggiore o familiarità per malattia depressiva, ma correlato ad un declino età-dipendente dei valori di testosterone (28). Gli uomini affetti da questo particolare disturbo sembrano presentare valori di testosterone più bassi dei controlli di pari età, non depressi, e la terapia sostitutiva sembra esercitare un effetto positivo sul loro tono dell’umore (30,31). I lavori epidemiologici condotti su ampie popolazioni che comprendevano soggetti di entrambi i sessi, non sono stati tuttavia in grado di dimostrare un’associazione tra livelli di testosterone e prevalenza di depressione (32). Analogamente, gli studi condotti su pazienti con una diagnosi di depressione che rispecchiava i criteri del DSM-5, non hanno osservato alcuna superiorità del testosterone rispetto al placebo, nel controllo della malattia, né hanno verificato alcun beneficio derivante dalla somministrazione dello stesso nei pazienti non responsivi alla terapia psichiatrica (33,34).
Anche per quanto riguarda la popolazione femminile, i dati della letteratura non sono sempre concordi. Alcuni studi sembrano suggerire che alcune condizioni cliniche caratterizzate da aumentata concentrazione di androgeni, come la sindrome dell’ovaio policistico, siano associate allo sviluppo di ansia e sintomi depressivi (35,36), mentre per alcuni ricercatori la relazione tra testosterone e depressione nelle donne è mediata dalla condizione menopausale (37).
In letteratura ci sono studi in donne con depressione maggiore che hanno osservato valori di testosterone totale più bassi rispetto ai soggetti di controllo (38), mentre in uno studio prospettico su 3302 pazienti durato 8 anni le concentrazioni più elevate di testosterone si associavano a maggiore gravità della problematica psichiatrica (39). Altri autori non hanno invece trovato alcuna associazione tra livelli di androgeni o SHBG e sintomi o disturbi depressivi (40).
BIBLIOGRAFIA
- Andersen ML, Alvarenga TF, Mazaro-Costa R, et al. The association of testosterone, sleep, and sexual function in men and women. Brain Res 2011, 1416: 80-104.
- Ciocca G, Limoncin E, Carosa E, et al. Is testosterone a food for the brain? Sex Med Rev 2016, 4: 15-25.
- Celotti F, Negri-Cesi P, Poletti A. Steroid metabolism in the mammalian brain: 5-alpha-reduction and aromatization. Brain Res Bull 1997, 44: 365–75.
- Nunez JL, Huppenbauer CB, McAbee MD, et al. Androgen receptor expression in the developing male and female rat visual and prefrontal cortex. J Neurobiol 2003, 56: 293-302.
- Hamann S, Herman RA, Nolan L, Wallen K. Men and women differ in amygdala response to visual sexual stimuli. Nat Neurosci 2004, 7: 411-6.
- Beyenburg S, Watzka M, Clusmann H, et al. Androgen receptor mRNA expression in the human hippocampus. Neurosci Lett 2000, 294: 25–8.
- Lorenz B, Garcia-Segura LM, DonCarlos LL. Cellular phenotype of androgen receptor-immunoreactive nuclei in the developing and adult rat brain. J Comp Neurol 2005, 492: 456–68.
- Morris RW, Purves-Tyson TD, Weickert CS, et al. Testosterone and reward prediction-errors in healthy men and men with schizophrenia. Schizophr Res 2015, 168: 649–60.
- Nguyen TV, McCracken J, Ducharme S, et al. Testosterone-related cortical maturation across childhood and adolescence. Cereb Cortex 2013, 23: 1424-32.
- Nguyen TV. Developmental effect of androgens in human brain. J Neuroendocrinol 2008, 30: e12486.
- Abel KM, Drake R, Goldstein JM. Sex differences in schizophrenia. Int Rev Psychiatry 2010, 22: 417–28.
- Owens SJ, Purves-Tyson TD, Webster MJ. Evidence for enhanced androgen action in the prefrontal cortex of people with bipolar disorders but not schizophrenia or major depressive disorders. Psychiatry Res 2019, 280: 112503.
- Riecher-Rossler A. Oestrogen, prolactin, hypotalamic-pituitary-gonadal axis and schizophrenic psychoses. Lancet Psychiatry 2017, 4: 63-72.
- Piacentino D, Kotzalidis GD, Del Casale A, et al. Anabolic-androgenic steroid use and psychopathology in athletes. A systematic review. Curr Neuropharmacol 2015, 13: 101–21.
- Clark AS, Henderson LP. Behavioral and physiological responses to anabolic-androgenic steroids. Neurosci Biobehav Rev 2003, 27: 413-36.
- Henderson LP, Jorge JC. Steroid modulation of GABAA receptors: CNS roles in reproduction, dysfunction and drug abuse. In: Advances in molecular and cell biology: molecular insights into ion channel biology in health and disease. Maue RA ed. Elsevier, Amsterdam, 2004, vol. 32: pp 217-49.
- Pomara C, Neri M, Bello S, et al. Neurotoxicity by synthetic androgen steroids: oxidative stress, apoptosis, and neuropathology: a review. Curr Neuropharmacol 2015, 13: 132-45.
- Ko Y-H, Jung S-W, Joe S-H, et al. Association between serum testosterone levels and the severity of negative symptoms in male patients with chronic schizophrenia. Psychoneuroendocrinology 2007, 32: 385–91.
- Ji E, Weickert CS, Lenroot R, et al. Endogenous testosterone levels are associated with neural activity in men with schizophrenia during facial emotion processing. Behav Brain Res 2015, 286: 338–46.
- Li J, Xiao W, Sha W, et al. Relationship of serum testosterone levels with cognitive function in chronic antipsychotic-treated male patients with schizophrenia. Asia Pac Psychiatry 2015, 7: 323–9.
- Aydogan U, Aydogdu A, Akbulut H, et al. Increased frequency of anxiety, depression, quality of life and sexual life in young hypogonadotropic hypogonadal males and impacts of testosterone replacement therapy on these conditions. Endocr J 2012, 59: 1099-105.
- Gagliano-Jucà T, Travison TG, Nguyen PL, et al. Effects of androgen deprivation therapy on pain perception, quality of life and depression in men with prostate cancer. J Pain Symptom Manage 2018, 55: 307-17.
- Thomas HR, Chen MH, D’Amico AV, et al. Association between androgen deprivation therapy and patient-reported depression in men with recurrent prostate cancer. Clin Genitourin Cancer 2018, 16: 313-7.
- Ng HS, Koczwara B, Roder D, Vitry A. Development of comorbidities in men with prostate cancer treated with androgen deprivation therapy: an Australian population-based cohort study. Prostate Cancer Prostatic Dis 2018, 21: 403-10.
- Ford AH, Yeap BB, Flicker L, et al. Prospective longitudinal study of testosterone and incident depression in older men: the health in men study. Psychoneuroendocrinology 2016, 64: 57–65.
- Elliot J, Kelly SE, Millar AC, et al Testosterone therapy in hypogonadal men: systematic review and network meta-analysis. BMJ Open 2017, 7: e015284.
- Walther A, Breidenstein J, Miller R. Association of testosterone treatment with alleviation of depressive symptoms in men. A systematic review and meta-analysis. JAMA Psychiatry 2019, 76: 31-40.
- Bhasin S, Seidman S. Testosterone treatment of depressive disorders in men. Too much smoke, not enough high-quality evidence. JAMA Psychiatry 2019, 76: 9-10.
- Snyder PJ, Bhasin C, Cunningham GR, et al; Testosterone Trial Investigators. Effects of testosterone treatment in older men. N Engl J Med 2016, 374: 611-42.
- Shores MM, Kivlahan DR, Sadak TI, et al. A randomized double-blind placebo-controlled study of testosterone treatment in hypogonadal older men with subthreshold depression (dysthymia or minor depression). J Clin Psychiatry 2009, 70: 1009-16.
- Seidman SN, Orr G, Raviv G, et al. Effects of testosterone replacement in middle-aged men with dysthymia: a randomized placebo-controlled clinical trial. J Clin Psychopharmacology 2009, 29: 216-21.
- Kische H, Pieper L, Venz J, et al. Longitudinal change instead of baseline testosterone predicts depressive symptoms. Psychoneuroendocrinology 2018, 89: 7-12.
- Seidman SN, Spatz E, Rizzo C, Roose SP. Testosterone replacement therapy for hypogonadal men with major depressive disorder: a randomized placebo-controlled clinical trial. J Clin Psychiatry 2001, 62: 406-12.
- Pope HG, Amiaz R, Brennan BP, et al. Parallel-group placebo controlled trial of testosterone gel in men with major depressive disorders displaying an incomplete response to standard antidepressant treatment. J Clin Psychopharmacol 2010, 30: 126-34.
- Cooney LG, Lee I, Sammel MD, Dokras A. High prevalence of moderate and severe depressive and anxiety symptoms in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod 2017, 32: 1075-91.
- Dokras A, Clfton S, Futterweit W, Wild R. Increased prevalence of anxiety symptoms in women with polycystic ovary syndrome: systematic review and meta-analysys. Fertil Steril 2012, 97: 225-30.
- Fiacco S, Walther A, Ehlert U. Steroid secretion in healthy aging. Psychoneuroendocrinology 2019, 105: 64-78.
- Oulis P, Masdrakis VG, Markianos M. Testosterone and dehydroepiandrosterone sulfate in female anxious and non-anxious major depression. Int J Psychiatry Clin Pract 2014, 18: 21–4.
- Bromberger JT, Schott L, Kravitz HM, et al. Longitudinal change in reproductive hormones and depressive symptoms across the menopausal transition. Results from the study of Women’s Health Across the Nation. Arch Gen Psychiatry 2010, 67: 598-607.
- Giltay EJ, van der Mast RC, Lauwen E, et al. Plasma testosterone and the course of major depressive disorder in older men and women. Am J Geriatr Psychiatry 2017, 25: 425–37.
Effetti endocrino-metabolici dei farmaci psichiatrici
Paola Sartorato
Medicina Interna, Montebelluna
(aggiornato al 10 settembre 2017)
METABOLISMO
Il trattamento con farmaci psichiatrici, in particolare alcuni anti-psicotici, può correlarsi all’insorgenza di complicanze metaboliche e cardio-vascolari; i pazienti trattati possono essere a maggior rischio di obesità, sindrome metabolica e diabete tipo 2.
Recentemente le società scientifiche coinvolte hanno evidenziato la necessità che questi pazienti vengano regolarmente monitorati anche per queste sequele legate alla terapia. Questi eventi avversi si osservano più frequentemente in una popolazione considerata vulnerabile, ovvero soggetti al primo episodio di schizofrenia, naive per altri farmaci psicotropi, bambini e adolescenti. Le malattie e i fattori di rischio cardiometabolici presentati dai pazienti psichiatrici derivano dall’interazione di una serie di fattori, quali background genetico, patologie sottostanti, stile di vita ed effetti dei farmaci psicotropi (1).
Gli effetti avversi metabolici secondari ai trattamenti anti-psicotici sono oramai noti, di diversa entità in relazione alla classe del farmaco e alle caratteristiche cliniche del paziente. Il clinico dovrebbe scegliere il farmaco con minor impatto sul profilo metabolico, minimizzando i rischi; i pazienti dovrebbero essere incoraggiati e coinvolti in progetti educazionali riguardanti alimentazione e attività fisica.
Obesità
Pazienti affetti da malattia psichiatrica sono di per sé maggiormente a rischio di obesità, fattori di rischio cardiovascolare e morbilità e mortalità a essi correlate, con un impatto significativo sulla qualità di vita. L’incremento ponderale è un noto effetto avverso di alcuni dei farmaci anti-psicotici utilizzati, sia in acuto che in mantenimento, e può presentarsi nel 15-72% dei pazienti schizofrenici; evidenze suggeriscono effetti simili anche in pazienti con disturbo bipolare (2).
Studi e meta-analisi descrivono un’eterogeneità di effetto sul peso corporeo, sia tra i diversi anti-psicotici che per uno stesso farmaco da soggetto a soggetto.
| Tabella 1 Rischio di incremento ponderale associato ai diversi anti-psicotici (3) |
|
| Minore | Amilsulpiride, aripiprazolo, asenapina, lurasidone e ziprasidone |
| Intermedio | Iloperidone, quetiapina, risperidone, paliperidone, sertindolo e zotepina |
| Alto | Clozapina, olanzapina, clorpromazina e tioridazina |
Poiché con ogni classe di questi farmaci è stato evidenziato un incremento ponderale rispetto al placebo, nessuno di questi si può considerare neutrale sul peso corporeo.
L’incremento del peso corporeo in corso di terapia anti-psicotica sembra svilupparsi in tre stadi:
- rapido incremento del peso entro i primi tre mesi di terapia;
- progressivo ma più lento incremento del peso entro l’anno;
- plateau con mantenimento del peso nel tempo.
Diversi fattori possono contribuire all’incremento del peso, quali caratteristiche demografiche, ambito di trattamento, trattamenti pregressi e concomitanti, dieta, attività fisica e BMI. Per clozapina e olanzapina sembra esserci una correlazione positiva tra incremento del peso corporeo e concentrazione plasmatica dei farmaci.
L’approvazione da parte dell’FDA di alcuni anti-psicotici di seconda generazione per il trattamento di disturbo bipolare e schizofrenia anche in bambini/adolescenti ha consentito di osservare un impatto importante sull’incremento del peso corporeo in questa categoria di pazienti (4). Disregolazioni dell’appetito, dell’introito di cibo e del senso di sazietà moderano e modulano l’incremento di peso in questi pazienti. Studi condotti su modelli animali e in parte riprodotti nell’uomo hanno evidenziato effetti degli psicofarmaci su neurotrasmettitori, peptidi e ormoni che regolano l’appetito e l’omeostasi energetica. In pazienti trattati con alcuni anti-psicotici di seconda generazione si è osservato un significativo incremento dei livelli di leptina. I dati riguardanti la ghrelina sono eterogenei: sembra che i valori al mattino a digiuno decrescano all’inizio del trattamento, per poi aumentare dopo l’esposizione cronica agli anti-psicotici. Nell’uomo non vi è dimostrazione che gli anti-psicotici si leghino a recettori ipotalamici tradizionalmente associati con la regolazione del peso corporeo. L’unico meccanismo comune a tutti gli anti-psicotici è il forte legame al recettore dopaminergico D2 (antagonismo, agonismo parziale); gli anti-psicotici presentano capacità di legame e affinità diverse ai recettori di serotonina, acetilcolina, istamina e noradrenalina. È stata evidenziata una correlazione tra la modulazione dell’attività del recettore dopaminergico D2 e il comportamento alimentare (2). Poiché anche la serotonina gioca un ruolo importante nella regolazione dell’introito di cibo e del senso di sazietà, è logico supporre che gli effetti sul peso corporeo siano legati anche alla modulazione di questo sistema, vista l’azione antagonista sul recettore 5-HT2c della maggior parte degli anti-psicotici di seconda generazione. È importante sottolineare che l’effetto di questi farmaci sul peso corporeo non è attribuito alla modulazione di un solo neurotrasmettitore, ma a un sistema complesso, in cui sembra svolgere un ruolo importante anche la farmacogenomica, in particolare per i sistemi dopaminergico e serotoninergico (5).
Per quanto riguarda il monitoraggio di questa complicanza, una consensus (6) raccomandava già nel 2004 il monitoraggio mensile di peso, BMI e circonferenza vita nei primi 3 mesi di trattamento anti-psicotico, in particolare nei pazienti con noti fattori di rischio.
Vi sono evidenze cliniche sull’efficacia del cambio di anti-psicotico in caso di incremento corporeo > 5% in corso di terapia. Nei pazienti a maggior rischio sono suggeriti programmi educazionali dietetici; alcuni studi hanno evidenziato anche l’efficacia della metformina associata agli anti-psicotici nel ridurre l’impatto negativo della terapia sul peso corporeo (7).
Sindrome metabolica
Il trattamento con farmaci anti-psicotici pone il paziente a maggior rischio di presentare anche altri eventi avversi cardiovascolari e metabolici, quali iperglicemia, ipertensione arteriosa e dislipidemia, maggiormente evidenti in corso di primo trattamento (8).
| Tabella 2 Rischio di sviluppare sindrome metabolica associato ai diversi anti-psicotici (3) |
|
| Minore | Aripiprazolo e ziprasidone |
| Intermedio | Quetiapina, risperidone, paliperidone, amisulpiride e sertindolo |
| Alto | Clozapina, olanzapina e clorpromazina |
Una meta-analisi condotta su 112 studi ha rilevato che il 49.7% dei pazienti in trattamento con clozapina presentava un quadro di sindrome metabolica. Circa il 25% dei pazienti in trattamento anti-psicotico ha alterata glicemia (> 100 mg/dL) e circa il 50% presenta dislipidemia (9). Il peggioramento dei valori pressori sembra secondario all’incremento del peso corporeo ed è in parte attenuato dall’azione ipotensiva di alcuni di questi farmaci, che agiscono come modulatori agonisti dei recettori adrenergici.
| Tabella 3 Rischio di dislipidemia associato ai diversi anti-psicotici (3) |
|
| Minore | Aripiprazolo, ziprasidone |
| Intermedio | Quetiapina, risperidone |
| Alto | Clozapina, olanzapina |
Gli effetti di olanzapina, clozapina e quetiapina non sono secondari solo all’incremento del peso, ma legati a un’azione diretta del farmaco sul metabolismo lipidico, che determina in particolare un incremento dei livelli dei trigliceridi, ma anche di colesterolo LDL. Non sono ancora chiari i meccanismi fisiopatologici sottostanti questa disregolazione del profilo lipidico.
Anche per questa possibile complicanza si suggerisce valutazione del profilo lipidico a inizio terapia, dopo 3 mesi e poi annualmente o dopo 5 anni in relazione al rischio (6). Come per l’obesità, vi sono evidenze sull’efficacia di un eventuale cambio di farmaco, scegliendone uno più neutro sull’assetto lipidico.
Diabete mellito
In corso di trattamento con anti-psicotici vi è un incremento del rischio di sviluppare DM tipo 2, in particolare per i farmaci di seconda generazione (1.3 volte vs farmaci di prima generazione). Il rischio è comunque individualizzato per tipo di farmaco: in particolare, olanzapina, clozapina e in misura minore quetiapina e risperidone, pongono il paziente a maggior rischio di sviluppare questa complicanza (3). Anche in questo caso i soggetti trattati giovani (bambini/adolescenti) sembrano a maggior rischio di alterazioni del metabolismo glucidico, in assenza, anche per età, di altri fattori di rischio associati. L’impatto di questi farmaci, in particolare clozapina e olanzapina, sul sistema glucidico sembra essere legato alla loro capacità di legame per i recettori muscarinici M2 e M3 espressi nel pancreas, con effetti sulla secrezione insulinica legata alla via colinergica (10).
ALTRE PATOLOGIE ENDOCRINE
Prolattina
L'iperprolattinemia secondaria all’assunzione di anti-psicotici/neurolettici è nota fin dai primi anni ‘70. L’iperprolattinemia in pazienti affetti da malattia psichiatrica è comune, in particolare nella schizofrenia la frequenza è elevata e correlata a tipo di farmaco e dosaggio in corso. Il 40-90% dei pazienti in trattamento con fenotiazine o butirrofenoni presenta iperprolattinemia, fino ad arrivare al 50-100% dei pazienti in terapia con risperidone (11).
Tutti i farmaci anti-psicotici hanno azione antagonista sul sottotipo recettoriale dopaminergico D2. Gli anti-psicotici tipici agiscono bloccando tutte le vie dopaminergiche cerebrali; alcuni atipici, in particolare clozapina, olanzapina, quetiapina e aripiprazolo, hanno un’azione antagonista più selettiva sulle vie dopaminergiche, con minor impatto sui livelli di PRL (12). L’aripiprazolo, agonista parziale del recettore D2, può addirittura ridurre l’iperprolattinemia. Non è comunque possibile categorizzare chiaramente gli anti-psicotici in base agli effetti sui livelli di prolattina, essendo questi correlati anche a dose, durata del trattamento, età e genere. Oltre ai neurolettici, anche alcuni anti-depressivi, come clomipramina e fluoxetina, possono determinare lieve iperprolattinemia, mentre benzodiazepine, litio e anti-convulsivanti hanno bassa probabilità di alterare i livelli di PRL.
Generalmente i livelli di iperprolattinemia secondari a farmaci variano tra 25-100 µg/L, ma risperidone e fenotiazine possono indurre valori anche > 200 µg/L. In presenza di iperprolattinemia in corso di trattamento con anti-psicotici, la sospensione o il cambio della terapia devono sempre essere attentamente discussi con lo psichiatra. Se il trattamento non può essere sospeso o modificato con farmaci con minor impatto sulla PRL e l’insorgenza dell’iperprolattinemia non può essere ricondotta con certezza all’inizio della terapia, è raccomandata la RMN ipofisaria per escludere patologie concomitanti della regione ipotalamo-ipofisaria, in particolare per valori significativi di iperprolattinemia.
Alcuni pazienti con iperprolattinemia farmaco-indotta possono rimanere asintomatici e non devono essere trattati, mentre in presenza di persistenti segni e sintomi di ipogonadismo, può essere presa in considerazione la terapia con estrogeni o testosterone. L’indicazione al trattamento con dopamino-agonisti in questa categoria di pazienti è controversa: gli studi riportano una percentuale di normalizzazione della PRL di circa il 75%, a scapito di un possibile aggravamento della psicosi sottostante; pertanto, anche questa indicazione dovrà essere attentamente discussa con lo psichiatra.
ACTH
Numerose evidenze sottolineano la complessa relazione tra stress, sia fisico che psichico, e alterazioni dell’asse ipotalamo-ipofisi-surreni. La valutazione delle modificazioni di questo asse nell’ambito della patologia psichiatrica è complicata, sia per le modalità di studio dell’asse che per la diverse fasi di malattia in cui questo può essere studiato. Valutazioni più accurate, anche se molto eterogenee, sulle modificazioni dei livelli di cortisolo e ACTH riguardano soprattutto i pazienti schizofrenici e affetti da depressione maggiore.
Nella schizofrenia i livelli di cortisolemia possono essere più elevati nei pazienti rispetto ai controlli in diverse fasi della malattia e con sintomatologia differente. Alcuni studi hanno inoltre evidenziato in alcuni di questi pazienti la scomparsa del ritmo circadiano di secrezione dell’ormone e in condizioni acute di stress fisico/psicologico una risposta ridotta sia di cortisolo che di ACTH ai test rispetto ai controlli, con un profilo di risposta differente rispetto ai pazienti affetti da sindrome depressiva. Escludendo interferenze farmacologiche, circa il 25% dei pazienti schizofrenici sembra avere livelli di cortisolo non sopprimibili dopo desametasone. Ci sono alcuni studi preliminari e non conclusivi sul possibile utilizzo di terapie di blocco dell’asse corticotropo (chetoconazolo, mifepristone) in pazienti affetti da psicosi.
In pazienti affetti da depressione maggiore, è frequente l’attivazione dell’asse corticotropo con mancata soppressione del cortisolo dopo desametasone e i trattamenti farmacologici possono contribuire a regolarizzare l’asse corticotropo, condizione considerata favorevole ma non sufficiente per valutare l’esito del trattamento.
I farmaci anti-psicotici possono influenzare i livelli di cortisolo in vari modi: migliorando la sintomatologia, possono ridurre lo stato di stress e quindi interferire sui livelli di cortisolemia; inoltre, possono influenzare direttamente la secrezione dell’ormone per effetto farmacologico diretto sull’asse. I livelli di cortisolo sembrano essere poco influenzati dagli anti-psicotici di prima generazione, mentre quelli di seconda generazione, in particolare olanzapina, quetiapina e clozapina, possono ridurre in modo significativo i livelli di ACTH e cortisolo, probabilmente attraverso le loro azioni serotoninergiche, adrenergiche e istaminergiche; tale riduzione farmaco-indotta di cortisolo e ACTH è stata documentata anche in soggetti sani (13).
Osteoporosi
La schizofrenia è associata a ridotta BMD, sia nei maschi che nelle femmine, con incrementato rischio di frattura. Rimane difficile chiarire quanto pesino su questa importante comorbilità la malattia di per sé, l’iperprolattinemia farmaco-indotta e i fattori di rischio legati alle abitudini di vita: ridotta attività fisica, fumo di sigaretta, abuso di alcolici, deficit di vitamina D sono fattori di rischio noti per osteoporosi, spesso presenti nella storia di questi pazienti. I farmaci anti-psicotici contribuiscono alla riduzione del BMD, sicuramente mediante l’ipogonadismo secondario all’iperprolattinemia, che pertanto dovrà essere trattato, mentre in questi pazienti le evidenze su iperprolattinemia di per sé e osso non sono ancora tali da suggerire raccomandazioni cliniche specifiche (14,15).
La relazione tra depressione e rischio maggiore di fratture è nota fin dagli anni ’90; evidenze più recenti hanno dimostrato una correlazione tra depressione e osteoporosi e una riduzione della BMD nei casi rispetto ai controlli. Anche in questi pazienti coesistono molteplici fattori di rischio più o meno noti per osteoporosi (ipercortisolismo, iponatremia), oltre all’assunzione di farmaci anti-depressivi. Studi più numerosi con dati sperimentali e clinici riguardano gli inibitori della ricaptazione della serotonina (SRI) e i triciclici e i loro effetti su osteoblasti, osteociti e osteoclasti che esprimono recettori per la serotonina. Gli studi clinici hanno evidenziato un’aumentata perdita di massa ossea, in particolare nelle donne in post-menopausa in corso di terapia con SRI (16).
Tiroide
Nell’ambito delle terapie psichiatriche sono ben noti gli effetti del litio su morfologia e funzionalità tiroidea. Il litio interferisce con la funzionalità tiroidea agendo a vari livelli: compete con il trasportatore dello iodio, inibisce la sintesi e la secrezione degli ormoni tiroidei, altera la conformazione e la funzione della tireoglobulina e riduce la desiodazione e la clearance della T4. L’anomalia tiroidea più frequente indotta dal litio è il gozzo, riscontrato in circa il 50% dei pazienti, prevalenza che varia in relazione all’area geografica, alla durata del trattamento e agli strumenti diagnostici usati.
L’ipotiroidismo subclinico e conclamato sono anch’essi molto frequenti, con frequenze variabili dal 10 al 50% circa, quest’ultima in particolare in pazienti con positività autoanticorpale. L’ipotiroidismo insorge in media dopo circa 18 mesi dall’inizio del trattamento, ma può comparire anche precocemente; sono a maggior rischio le donne, con storia familiare, con anticorpi positivi e > 50 anni. In pazienti suscettibili il litio incrementa il rischio di sviluppare una patologia autoimmune. Viene suggerito un controllo annuale della funzionalità tiroidea, da anticipare se presenti più fattori di rischio. L’approccio diagnostico e terapeutico, sia per il gozzo che per il distiroidismo è simile a quello della popolazione generale.
L’ipertiroidismo indotto da litio è molto meno frequente e può presentarsi sia come tiroidite non dolente e transitoria a ridotta captazione (pertanto non responsiva a trattamento radiometabolico), sia come forma autoimmune con positività auto-anticorpale (7).
Gli altri psico-farmaci incidono assai meno frequentemente sulla funzione tiroidea:
- la quetiapina è stata associata con un modesto incremento dei valori di T4 senza ripercussioni sui valori di TSH;
- fenitoina e carbamazepina possono ridurre il legame per la TBG e aumentare i livelli circolanti di ormone tiroideo libero;
- gli anti-depressivi triciclici possono interferire sull’asse ipotalamo-ipofisi-tiroide, riducendo la risposta del TSH al TRH. Per i pazienti in trattamento con questi ultimi farmaci non vi sono linee guida specifiche, ma è raccomandato un controllo periodico della funzionalità tiroidea (17).
Metabolismo del sodio
L’iponatremia rappresenta un disturbo elettrolitico frequente nei pazienti psichiatrici, la cui valutazione, a volte complessa, richiede una diagnosi differenziale tra una forma secondaria ai trattamenti in atto e una forma legata alla psicosi, con polidipsia psicogena e intossicazione d’acqua. Tra i farmaci psico-attivi la SIADH è stata associata all’assunzione di SRI, carbamazepina, oxcarbazepina e litio. La frequenza di iponatremia secondaria all’uso di SRI è variabile e legata al cut-off usato: Na < 135 mEq/L nel 9-40%, < 130 mEq/L nello 0.6-2.6%, con rischio maggiore nei pazienti con concomitante utilizzo di diuretici tiazidici. Gli SRI a maggior rischio sembrano essere fluoxetina, citalopram ed escitalopram, mentre sembra minore il rischio con paroxetina e sertralina. Anche duloxetina e venlafaxina possono indurre iponatremia precocemente, in particolare nei soggetti anziani. I triciclici e la mirtazapina hanno un minor profilo di rischio per l’iponatremia. Si tratta di una complicanza potenzialmente severa legata ai trattamenti, di cui bisogna tener conto, in particolare in soggetti anziani con concomitanti fattori di rischio che manifestino alterazioni dello stato di coscienza (18).
I sali di litio, ampiamente utilizzati nel trattamento del disturbo bipolare, rappresentano la più frequente causa iatrogena di diabete insipido (DI) nefrogenico: si stima che in corso di terapia con litio dal 20 al 40% dei pazienti possa presentare DI, con l’insorgenza di ridotta capacità di concentrare le urine e quindi moderata poliuria già a 8 settimane dall’inizio del trattamento, effetto generalmente reversibile alla sospensione della terapia. In alcuni casi sono richiesti mesi o anni per un recupero totale della capacità renale di concentrare le urine. Gli effetti renali del litio sono legati al dosaggio, alla durata del trattamento, all’età del paziente, alle comorbilità e al contemporaneo utilizzo di altri farmaci. Dopo un uso prolungato del farmaco, in alcuni pazienti si instaura un danno renale irreversibile da nefropatia interstiziale cronica, anche se il rischio assoluto sembra comunque basso rispetto alla popolazione generale (0.5% vs 0.2%). Il litio è un composto naturale, idrosolubile, che non si lega alle proteine plasmatiche e viene eliminato immodificato quasi esclusivamente dal rene. A livello renale il litio viene filtrato completamente a livello del glomerulo come sodio e potassio e viene riassorbito per circa l’80% a livello del tubulo prossimale; ne deriva che circa il 20% del litio filtrato si ritrova a livello del dotto collettore. All’aumento del litio intra-cellulare corrisponde a livello renale un’inibizione del cotrasporto Li-Na, con aumento dell’eliminazione del sodio dall’organismo e conseguente poliuria. L’accumulo di concentrazioni citotossiche di litio determina una ridotta espressione delle acquaporine, in particolare AQP2 e 3; la riduzione di tali canali dell’acqua, regolati dalla vasopressina (AVP) e localizzati a livello apicale della membrana plasmatica delle cellule dei dotti collettori, determina marcata inibizione del riassorbimento d’acqua e quindi poliuria e polidipsia e conseguente diabete insipido nefrogenico, definito dalla perdita di un volume urinario > 3-3.5 L/24h o > 50 mL/kg/24h, con osmolalità urinaria < 300 mOsm/L. Studi recenti hanno dimostrato come il DI nefrogenico indotto da litio non sarebbe secondario solo all’azione sulla via di segnale di AVP, ma sarebbero coinvolti anche altri fattori AVP-indipendenti, che contribuirebbero a determinare un danno del sistema tubulare (19). In molti pazienti l’aumentato introito di acqua è sufficiente per compensare la poliuria indotta dal farmaco. Nelle forme più severe può essere utilizzata l’amiloride, che inibendo il trasportatore del sodio (ENaC) riduce l’ingresso e quindi la concentrazione di litio nelle cellule, oltre che ridurre il volume urinario. Anche i tiazidici possono rappresentare un’opzione terapeutica, inducendo ipovolemia e determinando un incremento del riassorbimento tubulare di acqua, con riduzione della poliuria. Recentemente nel trattamento del DI nefrogenico da litio resistente ad altre terapie è stato segnalato l’utilizzo con successo di acetazolamide (20).
BIBLIOGRAFIA
- Correll CU, et al. Antipsychotic drugs and obesity. Trends Molec Med 2011, 17: 97-107.
- De Hert M, et al. Physical illness in patients with severe mental disorders. Prevalence, impact of medications and disparities in health care. World Psychatry 2011, 10: 52–77.
- De Hert M, et al. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol 2012, 8: 114–26.
- Ames D, et al. Detecting and managing adverse effects of antipsychotic medications: current state of play. Psychiatr Clin North Am 2016, 39: 275–311.
- Lencz T, Malhotra AK. Pharmacogenetics of antipsychotic-induced side effects. Dialogues Clin Neurosci 2009, 11: 405–15.
- American Diabetes Association, American Psychiatric Association, American Association of Clinical Endocrinologists, and North American Association for the Study of Obesity. Consensus development conference on antipsychotic drugs and obesity and diabetes. Diabetes Care 2004, 27: 596–601.
- Jarskog LF, Hamer RM, Catellier DJ, et al. Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective. Am J Psychiatry 2013, 170: 1032–40.
- Foley DL, Morley KI. Systematic review of early cardiometabolic outcomes of the first treated episode of psychosis. Arch Gen Psychiatry 2011, 68: 609–16.
- Mitchell AJ, et al. Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders — A systematic review and meta-analysis. Schizophr Bull 2013, 39: 306–18.
- Coccurello R, Moles A. Potential mechanisms of atypical antipsychotic-induced metabolic derangement: clues for understanding obesity and novel drug design. Pharmacol Ther 2010, 127: 210–51.
- Melmed S, Casanueva FF, Hoffman AR, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011, 96: 273-88.
- Wong-Anuchit C. Clinical management of antipsychotic-induced hyperprolactinemia. Perspect Psychiatr Care 2016, 52: 145-52.
- Bradley AJ, Dinan TG. A systematic review of hypothalamic-pituitary-adrenal axis function in schizophrenia: implications for mortality. J Psychopharmacol 2010, 24 (4 Suppl): 91-118.
- Kishimoto T, De Hert M, Carlson HE, et al. Osteoporosis and fracture risk in people with schizophrenia. Curr Opin Psychiatry 2012, 25: 415-29.
- Tseng PT, Chen YW, Yeh PY, et al. Bone mineral density in schizophrenia: an update of current meta-analysis and literature review under guideline of PRISMA. Medicine (Baltimore) 2015, 94: e1967.
- Rizzoli R, Cooper C, Reginster JY, et al. Antidepressant medications and osteoporosis. Bone 2012, 51: 606-13.
- Kibirige D, Luzinda K, Ssekitoleko R. Spectrum of lithium induced thyroid abnormalities: a current perspective. Thyroid Res 2013, 6: 3.
- De Picker L, Van Den Eede F, Dumont G, et al. Antidepressants and the risk of hyponatremia: a class-by-class review of literature. Psychosomatics 2014, 55: 536-47.
- Moeller HB, Rittig S, Fenton RA. Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment. Endocr Rev 2013, 34: 278-301.
- Gordon CE, Vantzelfde S, Francis JM. Acetazolamide in lithium-induced nephrogenic diabetes insipidus. N Engl J Med 2016, 375: 2008-9.
Forme congenite di tesaurismosi ed endocrinopatie
Carla Bizzarri
UOC Endocrinologia e Diabetologia, Ospedale Bambino Gesù, IRCCS, Roma
SI suddividono in malattie da accumulo di sostanze:
- normalmente presenti nell’organismo, ma prodotte o assorbite in quantità eccessiva, o rimosse in modo inadeguato (es. emocromatosi, malattia di Wilson, malattie lisosomiali);
- estranee all’organismo, introdotte dall’esterno e che non possono essere adeguatamente eliminate (es. carbone, asbesto).
In rapporto al tipo di metabolita o di organello cellulare coinvolto dal patologico accumulo di sostanza, si distinguono:
- malattie del metabolismo dei carboidrati;
- malattie del metabolismo degli aminoacidi;
- malattie del metabolismo dei lipidi;
- disordini congeniti della glicosilazione;
- malattie perossisomiali;
- malattie mitocondriali;
- malattie del metabolismo di purine e pirimidine;
- malattie del metabolismo intermedio;
- iperammoniemie;
- malattie del metabolismo di alcuni metalli;
- malattie da alterazione dei neurotrasmettitori;
- malattie lisosomiali:
- da deficit di degradazione delle glicoproteine;
- da alterato trasporto degli enzimi e dei substrati lisosomiali.
L'esempio più conosciuto di malattia da sovraccarico di una sostanza normalmente presente nell’organismo è l'emocromatosi ereditaria (primitiva), in cui il ferro è assorbito in quantità eccessiva per un difetto nel controllo del passaggio del ferro dalla cellula intestinale al sangue. Con il termine emocromatosi secondaria (o emosiderosi) si intende invece il sovraccarico di ferro associato a difetti nella produzione dei globuli rossi (anemie diseritropoietiche), epatopatie croniche di varia natura, somministrazione cronica di ferro per via parenterale (trasfusioni, preparati di ferro per via venosa o intramuscolare). La talassemia major e l'anemia sideroblastica sono i due esempi più studiati di sovraccarico di ferro secondario alla somministrazione ripetuta di trasfusioni di sangue e/o ad un'alterata produzione dei globuli rossi (eritropoiesi inefficace). Nella talassemia il ferro si accumula nei macrofagi e nel parenchima di fegato, pancreas, cuore e ghiandole endocrine, con conseguente fibrosi d’organo e insufficienza funzionale.
Le ghiandole endocrine maggiormente colpite dal danno da accumulo sono ipofisi, tiroide e pancreas endocrino, ma possono essere coinvolte anche paratiroidi e gonadi.
Le malattie lisosomiali
Il lisosoma è l’organello cellulare che ha la funzione di degradare proteine e strutture cellulari ormai senescenti, mediante una serie di enzimi, appartenenti alla categoria delle idrolasi acide. Le malattie lisosomiali conosciute sono circa 50, legate a diversi tipi di difetti genetici, accomunate dalla caratteristica di determinare un accumulo di metaboliti o sostanze nei lisosomi, con conseguente perdita di funzionalità cellulare. Le malattie lisosomiali sono classificate generalmente in base alla natura del materiare patologico accumulato, e possono essere suddivise secondo la classificazione ICD-10 in:
- (E75) malattie da accumulo lipidico (es. sfingolipidosi, come la malattia di Gaucher e la malattia di Niemann-Pick);
- (E75.0-E75.1) gangliosidosi (es. malattia di Tay-Sachs);
- (E75.2) leucodistrofia;
- (E76.0) mucopolisaccaridosi (inclusa la sindrome di Hunter e la malattia di Hurler);
- (E77) disordini da accumulo di glicoproteine;
- (E77.0-E77.1) mucolipidosi.
Si tratta di malattie sistemiche che si trasmettono in generale con modalità autosomica recessiva. L’età d’esordio è variabile, ma in genere si manifestano durante la prima infanzia. Le manifestazioni cliniche più tipiche sono rappresentate da:
- epatosplenomegalia;
- interessamento del sistema nervoso centrale, con ritardo dello sviluppo neuromotorio, o perdita progressiva di funzioni neurologiche già acquisite;
- alterazioni di occhi, cuore e muscolatura.
Nessun trattamento si è finora rivelato del tutto risolutivo per queste patologie. Tentativi terapeutici sono stati effettuati mediante:
- terapia sostitutiva enzimatica, attraverso la somministrazione periodica della forma normale dell'enzima mutato;
- trapianto di midollo;
- somministrazione di farmaci che riducono la sintesi delle sostanze che non sono efficacemente degradate nei lisosomi;
- stimolazione dell'attività enzimatica residua, attraverso farmaci che stabilizzano l'enzima mutato.
Per approfondimenti e ricerca dei Centri di Riferimento per le diverse patologie vedi Orphanet (portale delle malattie rare e dei farmaci orfani)
Emoglobinopatie
Patrizia Del Monte1, Marina Baldini2 & Maurizio Poggi3
1Genova
2Medicina Generale a Indirizzo Metabolico, Ospedale Maggiore Policlinico Ca’ Granda, Milano
3UOC Endocrinologia, AO Sant’Andrea, Roma
(aggiornato al 2024)
Introduzione
Nel 2022 sono state pubblicate le Buone Pratiche Cliniche nelle Complicanze Endocrinologiche delle Emoglobinopatie, promosse dalla SITE (Società Italiana Talassemie ed Emoglobinopatie) (1).
Il panel era composto da ematologi ed endocrinologi esperti in emoglobinopatie, affiancati da esperti con competenze metodologiche e organizzative. Sono state prese in considerazione le linee guida (LG) già esistenti per le diverse endocrinopatie nella popolazione generale, adattandole ai pazienti con emoglobinopatie e formulando raccomandazioni specifiche per questa popolazione. Il documento, disponibile anche nella versione italiana (2), comprende le raccomandazioni formulate secondo il sistema GRADE (a favore o contro; forte = si raccomanda; debole = si suggerisce) e la discussione delle loro motivazioni.
Le endocrinopatie rappresentano oggi le complicanze più frequenti nei pazienti con emoglobinopatie, in particolare nei pazienti affetti da talassemia trasfusione-dipendente, con un importante impatto sulla qualità di vita. L’accumulo di ferro ne è la causa principale ed è difficile sospettarle in base alla comparsa di segni e sintomi clinici, che spesso sono comuni alla malattia di base. Pertanto viene proposto un monitoraggio attivo (esame clinico e screening laboratoristico), per permetterne il pronto riconoscimento e trattamento, anche per ottimizzare la terapia ferro-chelante.
L’endocrinologo deve fare parte del team di cura, per il monitoraggio, il riconoscimento e la gestione precoce e continuativa delle complicanze endocrine delle emoglobinopatie (raccomandazione – R – 1) in tutte le fasi della vita.
Ipostaturismi (R 2-8)
Nei pazienti talassemici è frequente la bassa statura, spesso disarmonica, con crescita predominante degli arti e brevità del tronco.
Riconosce una genesi multi-fattoriale, con meccanismi che comprendono l’anemia cronica, il sovraccarico di ferro, la tossicità dei farmaci ferro-chelanti (in particolare della deferoxamina), la plati-spondilia; a questi si possono sovrapporre gli effetti negativi di eventuali deficit endocrini (ipotiroidismo, alterazione dell’omeostasi glucidica e calcio-fosforica, ritardo puberale/ipogonadismo, deficit di GH - GHD).
Lo screening (clinico e auxologico) è raccomandato ogni 6 mesi a partire dalla presa in carico del paziente, fino al completamento dello sviluppo staturale e puberale. Nei pazienti con rallentamento della crescita/deficit staturale si raccomanda di verificare il controllo ottimale della terapia trasfusionale e chelante e lo stato nutrizionale, di eseguire lo screening per celiachia e di correggere altre eventuali patologie endocrine individuate. Se i primi accertamenti sono negativi, si raccomandano dosaggio di IGF-I e test dinamici per GH (a seconda dell’esperienza del centro e della disponibilità dei test, evitando l’ipoglicemia insulinica per i rischi cui può esporre pazienti già fragili). I criteri diagnostici, l’eventuale trattamento con rhGH ricombinante e il suo monitoraggio sono del tutto sovrapponibili a quelli indicati nelle LG per la diagnosi e il trattamento del GHD nell’infanzia e nell’adolescenza per la popolazione generale e nella nota AIFA n. 39.
Prima dell’inizio della terapia con rhGH, si raccomanda di valutare metabolismo glucidico, funzionalità tiroidea e ipofisaria (ACTH, cortisolo, FT4 e TSH, in età puberale LH, FSH, testosterone totale/estradiolo) e di eseguire RM della regione ipotalamo-ipofisaria.
Follow-up: nei soggetti in terapia con rhGH, si raccomanda di rivalutare ogni 6 mesi i parametri auxologici, IGF-1, glicemia, annualmente età ossea, profilo lipidico, le altre tropine ipofisarie e la funzionalità tiroidea. Nei pazienti trattati con GH dall’età infantile si raccomanda la ripetizione del test dinamico in età di transizione, dopo almeno un mese di sospensione della terapia con GH, per valutare la necessità di proseguire o meno la terapia, a un dosaggio appropriato per l’età. Fanno eccezione i pazienti con panipopituitarismo, quelli con malformazioni congenite della regione ipotalamo-ipofisaria e i portatori di mutazioni genetiche che comportano un GHD, in cui il re-testing non è necessario.
Disturbi dello sviluppo puberale (R 9-18)
Rappresentano la più frequente problematica endocrinologica nel paziente pediatrico con talassemia major.
Hanno un disturbo dello sviluppo puberale:
- i ragazzi con assenza di segni di sviluppo puberale (volume testicolare < 4 mL) dopo i 14 anni o con mancata progressione puberale entro un periodo di 6-12 mesi dopo l’inizio spontaneo della pubertà;
- le ragazze con assenza di telarca dopo i 13 anni o con mancata progressione puberale dopo l’inizio spontaneo della pubertà, o con mancata comparsa di menarca dopo 4 anni dalla comparsa del telarca, o con amenorrea primaria entro i 16 anni di età.
Lo screening dei disturbi dello sviluppo puberale, raccomandato con cadenza semestrale a partire dai 12 anni fino a completamento dello sviluppo, include la valutazione dello stadio di Tanner, della velocità di crescita e del volume testicolare nel maschio. In caso di ritardo puberale, si raccomandano i dosaggi di LH, FSH, 17ß-estradiolo nelle femmine/testosterone totale nei maschi, TSH, FT4, prolattina, IGF-1, la valutazione dell'età ossea e l’ecografia pelvica nella femmina.
In tutti i pazienti con mancato inizio di sviluppo puberale, per discriminare il ritardo costituzionale di crescita e sviluppo puberale dall’ipogonadismo ipogonadotropo, si raccomanda un breve ciclo di 3-6 mesi di terapia ormonale a basse dosi (estrogeni nella femmina e testosterone nel maschio), seguito da un periodo di sospensione con rivalutazione clinico-laboratoristica. In caso di ipogonadismo ipogonadotropo (la forma più frequente di ipogonadismo in questi pazienti), si raccomanda di eseguire anche RM ipofisaria.
Nei maschi con confermato ipogonadismo ipogonadotropo si raccomanda di aumentare gradualmente la dose di testosterone, fino al raggiungimento della dose dell’adulto nell’arco di 3-4 anni, controllando la testosteronemia totale per verificare/modulare il dosaggio del farmaco e monitorando la risposta alla terapia attraverso la valutazione auxologica (stadio puberale, crescita staturale) almeno ogni 3-6 mesi e dell’età ossea una volta all’anno. Inoltre in questi pazienti, come alternativa al testosterone solo per l’induzione dello sviluppo testicolare, si suggerisce di prendere in considerazione l’utilizzo di gonadotropine a partire dall'età di 14 anni.
Nelle femmine con ritardo puberale, la terapia raccomandata è il 17β-estradiolo a basse dosi, preferibilmente per via trans-dermica, aggiungendo la terapia con progestinico alla comparsa del flusso mestruale indotto oppure dopo 24-36 mesi dall’inizio della terapia con estrogeni, monitorando la risposta alla terapia attraverso la valutazione dello stadio puberale, della crescita staturale, delle dimensioni di utero e ovaie all’ecografia almeno ogni 3-6 mesi e dell’età ossea una volta l’anno.
Ipogonadismo nella donna adulta (R 19-28)
L’ipogonadismo ipogonadotropo è la complicanza endocrina più frequente nelle pazienti trasfusione-dipendenti e la migliore prevenzione è una precoce ed efficace ferro-chelazione.
Come modalità di screening, si raccomanda la valutazione anamnestica della storia mestruale almeno ogni 6 mesi. In caso di amenorrea/oligomenorrea, sono indicate la valutazione basale della funzione gonadica (FSH, LH, estradiolo) e l’ecografia pelvica; si raccomanda di escludere altre cause di oligo-amenorrea (quali iperprolattinemia, iperandrogenismo, ipotiroidismo e gravidanza in corso). In presenza di ipogonadismo ipogonadotropo, si consiglia l’esecuzione di RM ipofisaria e il controllo degli altri assi ormonali, con i dosaggi di ACTH e cortisolo al mattino, TSH, FT4, prolattina, IGF-1.
Si raccomanda di iniziare la terapia sostitutiva con ormoni gonadici (TOS) nelle pazienti con ipogonadismo in età pre-menopausale, dopo accurata valutazione dei fattori di rischio ed esclusione di controindicazioni (anche in accordo con la check-list AIFA per i prescrittori). Se presenti fattori di rischio trombofilico, si suggerisce la valutazione da parte dello specialista in coagulopatie. Nelle donne adulte in età pre-menopausale con ipogonadismo si suggerisce TOS utilizzando estradiolo, preferibilmente per via trans-dermica, associato a progestinico ciclico sequenziale; nelle donne isterectomizzate non è necessario associare il progestinico.
Nelle pazienti in terapia, si raccomanda il monitoraggio con l’esame obiettivo, la misurazione della pressione arteriosa e l’esecuzione di esami ematochimici (in particolare funzionalità epatica, colesterolo, HDL, trigliceridi, glicemia, creatininemia), da eseguire dopo il primo mese di terapia e poi trimestralmente/semestralmente, salvo diversa indicazione clinica. Si suggeriscono visita ginecologica ed ecografia pelvica annuali, salvo diversa indicazione. In caso di sanguinamento vaginale imprevisto, va esclusa una patologia uterina. Le donne vanno invitate a eseguire lo screening per il carcinoma mammario appropriato per l’età. Il trattamento va protratto fino all’età fisiologica della menopausa, salvo insorgenza di controindicazioni. In caso di concomitante terapia per ipotiroidismo, si raccomanda controllo di FT4 e TSH dopo 3 mesi dall’inizio della terapia estro-progestinica.
Infertilità femminile (R 29-38)
Le raccomandazioni riguardano gli aspetti diagnostici e terapeutici, che vanno dalla stimolazione ovarica alle tecniche di riproduzione assistita, da gestire in centri multi-disciplinari altamente specializzati, con esperienza specifica nella paziente con emoglobinopatia.
lpogonadismo nel maschio adulto (R 39-48)
Si suggerisce lo screening per ipogonadismo almeno una volta l’anno, mediante la valutazione anamnestica, clinica e il dosaggio di testosterone, LH e FSH. Hanno ipogonadismo i pazienti con segni e sintomi suggestivi, associati a bassi valori di testosterone, confermati in due determinazioni su prelievo effettuato con le corrette modalità. In presenza di ipogonadismo ipogonadotropo (la forma più frequente nel talassemico), si raccomanda il controllo dei restanti assi ormonali e della prolattina. L’ipogonadismo ipergonadotropo è raro in questa popolazione e pone indicazione all’esecuzione di ecografia testicolare (ed eventuali ulteriori accertamenti).
In assenza di controindicazioni, si raccomanda di iniziare la terapia sostitutiva con testosterone enantato o propionato oppure testosterone in gel somministrato per via cutanea, a dosaggi da adattare in base alla risposta clinica e ormonale. Nel paziente in età fertile si raccomanda di eseguire lo spermiogramma prima di iniziare la terapia con testosterone. Si raccomanda di non iniziare terapia con testosterone nei pazienti:
- che programmano una paternità a breve distanza (6-12 mesi successivi);
- con nodulo/massa prostatica palpabile, PSA > 4 ng/mL o > 3 ng/mL in presenza di elevato rischio di carcinoma prostatico (parente di primo grado con carcinoma prostatico), carcinoma prostatico, sindrome da apnee ostruttive del sonno grave non trattata, gravi disturbi ostruttivi a carico delle vie urinarie inferiori, scompenso cardiaco, carcinoma mammario, ictus recente, infarto miocardico acuto recente (6 mesi), fattori di rischio trombotico (in quest’ultimo caso si suggerisce valutazione da parte di specialista in coagulopatie).
Follow-up: si raccomanda il dosaggio di testosterone ogni 3 mesi fino al raggiungimento di valori normali, poi ogni 6 mesi. Negli ipogonadici in trattamento con testosterone, a partire dai 40 anni si raccomanda di monitorare il rischio di carcinoma prostatico in conformità con le LG specifiche.
Infertilità maschile (R 49-56)
Hanno infertilità i pazienti maschi che non hanno concepito dopo almeno 12 mesi di rapporti mirati non protetti e presentano spermiogramma alterato.
Si raccomanda di eseguire lo screening per infertilità nei pazienti adulti che desiderano la paternità e nei pazienti che non hanno ottenuto il concepimento dopo 12 mesi di rapporti regolari non protetti. Si raccomanda di includere nello screening iniziale per l'infertilità maschile: anamnesi generale e riproduttiva, esame obiettivo e spermiogramma secondo le indicazioni metodologiche OMS; se il primo spermiogramma presenta anomalie, si raccomanda di ripeterlo almeno una volta a distanza di 2-3 mesi. Si suggerisce l’esecuzione di ecografia testicolare in tutti i pazienti con infertilità. Si raccomanda il dosaggio di FSH, LH, testosterone e prolattina in tutti i pazienti con alterazione seminale.
Nei pazienti in terapia sostitutiva si raccomanda di sospendere il testosterone prima di iniziare la terapia di induzione della spermatogenesi, e di controllare ogni 3-6 mesi testosterone e spermiogramma in corso di terapia con gonadotropine. Si suggerisce valutazione dello specialista urologo esperto in andrologia o di un endocrinologo esperto in sessualità e procreazione in tutti i pazienti in fase di inquadramento diagnostico, nei pazienti con azoospermia ostruttiva, nei pazienti che necessitano di biopsia testicolare e in quelli destinati alle metodiche di procreazione assistita con estrazione testicolare di spermatozoi.
Alterazioni del metabolismo glucidico (R 57-63)
La prevalenza delle anomalie dell’omeostasi glucidica è aumentata nei pazienti con talassemia.
Il diabete correlato all’accumulo di ferro ha una fisiopatologia specifica: come nel diabete mellito di tipo 1, si riscontra deficit insulinico, che tuttavia di norma è relativo e non assoluto; come nel diabete mellito di tipo 2, l’esordio è in genere graduale e insidioso ed è frequente la resistenza all’insulina.
La diagnosi precoce delle alterazioni del metabolismo glucidico è critica, perché nelle fasi iniziali una terapia chelante ottimale ne può determinare la regressione. Le definizioni adottate per la diagnosi di diabete, di alterata glicemia a digiuno e di ridotta tolleranza al glucosio sono le stesse formulate dalle LG utilizzate nella popolazione generale, con l’esclusione della valutazione della emoglobina glicata (HbA1c), inattendibile in questa popolazione. Si raccomanda di eseguire lo screening per disturbi del metabolismo glucidico a partire dai 10 anni, ogni 2 anni fino ai 18 anni, in seguito ogni anno. Si raccomanda di eseguire carico orale di glucosio (OGTT) nei pazienti con alterata glicemia a digiuno; a completamento diagnostico, nei pazienti con diagnosi di diabete mellito si raccomanda il dosaggio plasmatico del C-peptide. Si suggerisce il calcolo dell’indice HOMA-IR come parametro di insulino-resistenza.
Per le donne in gravidanza, si raccomanda di eseguire lo screening per il diabete gestazionale con glicemia a digiuno mensilmente dall’inizio della gravidanza e OGTT a 24–28 settimane di gestazione, seguendo i criteri specifici nella popolazione generale, salvo diversa indicazione.
In presenza di un disturbo del metabolismo glucidico, si raccomanda di intensificare la terapia ferro-chelante e di modificare lo stile di vita. Nei pazienti con pre-diabete (alterata glicemia a digiuno e ridotta tolleranza al glucosio), si suggerisce di valutare la terapia con metformina, soprattutto in presenza di insulino-resistenza. Nei pazienti con diabete, si suggerisce di valutare la terapia con metformina o insulina in base alla presenza di sintomi, segni catabolici, valori di glicemie e di C-peptide. Lo schema raccomandato di terapia insulinica è il basal/bolus multi-iniettivo. La terapia insulinica con micro-infusore è raccomandata in pazienti selezionati: quelli che non riescono a raggiungere una buona gestione del diabete nonostante la terapia multi-iniettiva intensiva e ottimizzata e/o con ipoglicemia grave o notturna, in bambini < 2 anni, in età pediatrica in caso di elevata insulino-sensibilità, in pazienti con compromissione dello stile di vita con la terapia multi-iniettiva. Al momento non disponiamo di dati sufficienti sull’utilizzo dei nuovi farmaci (inibitori di SGLT-2 e agonisti GLP-1) nei talassemici diabetici, aspetto meritevole di studi appropriati in futuro.
Nei pazienti in terapia insulinica si raccomanda l'educazione all’auto-monitoraggio della glicemia (e dei chetoni se glicemia > 250 mg/dL). Si raccomanda di eseguire il monitoraggio continuo della glicemia nei pazienti con insufficiente controllo glicemico, persistente nel tempo, e/o con ipoglicemie gravi. Si raccomanda un’appropriata istruzione su prevenzione, riconoscimento e trattamento dell’ipoglicemia. Si suggerisce di monitorare periodicamente la fruttosamina. Si raccomanda di ricercare le complicanze micro- e macro-angiopatiche, come indicato nelle LG sul diabete nella popolazione generale.
lpotiroidismo (R 64-73)
Le definizioni di ipotiroidismo subclinico, ipotiroidismo primario franco e ipotiroidismo centrale sono le stesse indicate nelle LG per la popolazione generale.
La fisiopatologia dell’ipotiroidismo nella talassemia è legata al sovraccarico di ferro, mentre gli anticorpi anti-Tg e anti-TPO sono solitamente negativi.
L’ipotiroidismo primario (da accumulo di ferro a livello tiroideo) è il tipo prevalente; le forme subcliniche sono oggi le più frequenti, ma possono progredire nel tempo in conclamate. La presenza di ipotiroidismo primario franco nei pazienti talassemici si è ridotta negli ultimi decenni, facendosi meno frequente nei pazienti giovani; la riduzione correla significativamente con il declino della ferritina, a conferma dell’effetto favorevole di un precoce inizio della terapia ferro-chelante.
L’ipotiroidismo centrale, secondario ad accumulo di ferro in sede ipotalamo-ipofisaria, è più raro, anche se in crescente riconoscimento, a suggerire che un danno dell’asse TRH-TSH può rendersi evidente nel corso del tempo, in pazienti con età maggiore e livelli di ferritina più bassi rispetto a quelli che hanno già sviluppato ipotiroidismo primario.
Si suggerisce di iniziare lo screening per l’ipotiroidismo non oltre i 9 anni di età e si raccomanda di effettuarlo mediante il dosaggio di TSH e di FT4 da ripetere annualmente (semestralmente in pazienti con parametri di ferro non ottimali). Si suggerisce di eseguire lo screening per ipotiroidismo nelle donne che ricercano una gravidanza o hanno in programma una fertilizzazione assistita, e di ripeterlo mensilmente in gravidanza. Si raccomanda di eseguire lo screening per ipotiroidismo trimestralmente nei pazienti in terapia con farmaci interferenti con la funzionalità tiroidea, in particolare amiodarone. Alla diagnosi di ipotiroidismo primario si suggerisce l’esecuzione di ecografia tiroidea, da ripetere in base all’indicazione clinica. Nell’ipotiroidismo centrale si raccomanda di eseguire anche il controllo degli altri assi ipofisari e della RM ipotalamo-ipofisaria.
In caso di ipotiroidismo franco (sia primario che centrale), si raccomanda di iniziare sempre la terapia sostitutiva, previo controllo della funzionalità dell’asse ipofisi-surrene con compenso di un eventuale deficit di cortisolo accertato. Nel paziente con ipotiroidismo subclinico si suggerisce di valutare l’inizio della terapia caso per caso, in base ad età, livelli di TSH, quadro clinico, ma si raccomanda il trattamento nella paziente che ricerca una gravidanza o durante la gravidanza. Come terapia di scelta si raccomanda la L-tiroxina, da iniziare a dosi basse, particolarmente nei pazienti cardiopatici, fragili, con aumento graduale del dosaggio fino a target. È necessario evitare il sovra-dosaggio di T4, per l’aumentato rischio aritmico (in particolare di fibrillazione atriale) e di peggioramento dell’osteoporosi, entrambi spesso presenti in questo particolare ambito clinico.
Follow-up: nei pazienti con ipotiroidismo primario in terapia sostitutiva si raccomanda di eseguire dosaggio di TSH e FT4 ogni 6 settimane fino a normalizzazione del TSH, poi ogni 6 mesi; in corso di terapie interferenti con il metabolismo degli ormoni tiroidei, si suggerisce controllo di TSH e FT4 ogni 3 mesi. Nei pazienti con ipotiroidismo centrale in trattamento con L-tiroxina, si raccomanda di eseguire dosaggio di FT4 ogni 4 settimane fino al raggiungimento di un valore di FT4 nella norma, successivamente almeno ogni 6 mesi.
In gravidanza, il dosaggio di L-tiroxina va aumentato per sopperire all’aumentato fabbisogno, già a partire dalla IV settimana, e quindi adeguato in base agli esami, facendo controlli mensili di FT4 e TSH lungo tutto l’arco della gravidanza. Dopo il parto, il fabbisogno di L-tiroxina si riduce, per cui si riduce il dosaggio della terapia a livelli pre-concezionali e poi si adegua in base ai valori di TSH e FT4, eseguiti dopo circa 6 settimane dal parto.
Ipoparatiroidismo (R 74-80)
Hanno ipoparatiroidismo i pazienti con ipocalcemia (calcemia corretta per albumina < 8 mg/dL) in associazione a valori di paratormone ridotti o inappropriatamente normali, in almeno due valutazioni.
Esiste una grande eterogeneità nei dati di prevalenza di questa complicanza.
La fisiopatologia dell’ipoparatiroidismo nelle emoglobinopatie trasfusione-dipendenti è multi-fattoriale, ma il sovraccarico di ferro gioca il ruolo predominante.
Si suggerisce di eseguire lo screening per ipoparatiroidismo annualmente dai 10 anni di età, mediante dosaggio della calcemia corretta per albumina e della fosfatemia. Nei pazienti con ipocalcemia cronica (calcemia corretta per albuminemia < 8 mg/dL), si raccomanda di controllare i seguenti parametri sierici: paratormone, calcemia corretta, fosfato, magnesio, creatinina e 25OH-vitamina D, oltre alla calciuria delle 24 h.
Si raccomanda di iniziare la terapia in pazienti con sintomi da ipocalcemia e/o livelli di calcemia corretta per albuminemia < 8.0 mg/dL. Si raccomanda di utilizzare calcitriolo (a dosaggi variabili da 0.25 a 2 µg/die, da dividere in due somministrazioni orali) e calcio carbonato o calcio citrato (in media 1-2.5 g/die per via orale nell’adulto, frazionato in 2-3 somministrazioni). Il calcio citrato è preferibile al carbonato in caso di effetti collaterali, terapie con inibitori di pompa protonica o se è presente acloridria. Si suggerisce di associare supplementazione con vitamina D orale alla dose di 400-800 UI/die, tale da mantenere il dosaggio della 25OH vitamina D ai limiti inferiori della norma. Nei casi di ipoparatiroidismo cronico grave, non compensato dalla terapia convenzionale, in assenza di controindicazioni si suggerisce di valutare l’eventuale l’uso di analoghi del PTH, ma non sono disponibili studi specifici nei talassemici.
Nei pazienti con ipoparatiroidismo stabile, si raccomanda di eseguire monitoraggio clinico (valutazione dei sintomi da ipo/ipercalcemia) e di calcemia corretta per albuminemia, fosfato, magnesio, creatininemia e filtrato glomerulare stimato ogni 3-6 mesi, calciuria e creatininuria (urine delle 24 ore) annualmente. All’inizio della terapia o nel caso di modifiche del dosaggio di calcio e vitamina D, oppure nei casi di scarsa compliance alla terapia, si raccomanda di eseguire valutazione biochimica più ravvicinata, con cadenza settimanale o mensile. Si raccomanda di mantenere la calcemia ai limiti inferiori della norma, fosfatemia ai limiti alti della normalità, un prodotto calcemia x fosfatemia (misurati entrambi in mg/dL) < 55 mg2/dL2 (4.4 mmol2/L2), evitando ipercalciuria, nefrolitiasi e nefrocalcinosi. Si raccomanda di eseguire valutazione renale e delle vie urinarie in occasione del controllo ecografico addominale previsto per i pazienti con emoglobinopatia.
L’ipocalcemia acuta è una complicanza grave dell’ipoparatiroidismo franco. Va trattata in maniera opportuna, inizialmente con calcio gluconato ev diluito in fisiologica o glucosata 5%, embricando calcio e calcitriolo per via orale, guidati da uno stretto monitoraggio della calcemia, fino a passare alla sola terapia orale con calcio e calcitriolo quando i livelli sierici si sono normalizzati.
I pazienti emoglobinopatici possono presentare comorbilità associate, che spesso richiedono terapie multiple, potenzialmente gravate da effetto ipocalcemizzante. L’utilizzo di bisfosfonati endovenosi o di denosumab sottocutaneo, potenti anti-riassorbitivi ossei, può causare ipocalcemia soprattutto nei soggetti con deficit di vitamina D. Inoltre, gli inibitori di pompa protonica e alcuni anti-convulsivanti possono interferire con i valori di calcemia e di vitamina D.
Iposurrenalismo (R 81-89)
Nella popolazione con emoglobinopatia sono riportati tassi di prevalenza dell’iposurrenalismo estremamente variabili nelle diverse casistiche e influenzati dalle differenze nell’efficacia delle terapie chelanti e dalla modalità di diagnosi. I cut-off per la diagnosi di ipocortisolismo sono stati recentemente modificati, tenendo conto della maggiore sensibilità degli attuali metodi di dosaggio.
Si suggerisce di eseguire lo screening per iposurrenalismo negli adulti con storia di accumulo di ferro e altri deficit endocrinologici. La modalità di screening raccomandata comprende la valutazione clinica, il dosaggio di sodio e potassio, dell’ACTH e della cortisolemia alle ore 8 del mattino. Si suggerisce di eseguire il test di stimolo con ACTH nei pazienti con valori confermati di cortisolo sierico < 10 µg/dL (275 nmol/L). Nei pazienti con valori confermati di cortisolemia tra 10 e 15 µg/dL, si suggerisce di valutare l’esecuzione del test di stimolo in funzione del quadro clinico e anamnestico e della coesistenza di altri deficit della funzione ipotalamo-ipofisaria. I test di stimolo raccomandati sono il test all’ACTH, a bassa dose (1 µg ev) o a dose standard (250 µg ev), e il test al glucagone, da scegliere in base alle risorse e all’esperienza del centro, alla disponibilità dei farmaci e alla necessità di testare altri assi ormonali (possibile con test al glucagone). Hanno iposurrenalismo i pazienti con valori di cortisolemia basale alle ore 8 del mattino < 3 µg/dL (80 nmol/L) e/o picco del cortisolo < 15-18 µg/dL (400-500 nmol/L) ai test di stimolo. L’iposurrenalismo può invece essere escluso nei pazienti con valori di cortisolemia al mattino > 15 µg/dL (400 nmol/L). Hanno iposurrenalismo primario i pazienti con cortisolo ridotto e ACTH elevato. In presenza di iposurrenalismo primario, si suggerisce di eseguire imaging surrenalico (TC o RM). Hanno iposurrenalismo centrale i pazienti con cortisolo ridotto e ACTH ridotto o inappropriatamente normale (in assenza di farmaci interferenti). In presenza di iposurrenalismo secondario, si raccomanda l’esecuzione di RM ipofisaria e il controllo dei restanti assi ormonali (FSH, LH, FT4, TSH, prolattina, IGF-1, testosterone nel maschio ed estradiolo nella femmina).
Si raccomanda di iniziare la terapia cortisonica sostitutiva in tutti i pazienti con iposurrenalismo confermato. La terapia consiste nella somministrazione di glucocorticoidi, cercando di riprodurre la fisiologica produzione surrenalica, caratterizzata da una maggiore quota al mattino, con progressiva riduzione nella giornata. Negli adulti si utilizza una dose giornaliera orale di circa 15–25 mg di idrocortisone o 20–35 mg di cortisone acetato, frazionata in 2-3 somministrazioni, la maggiore al mattino. Non c’è esperienza sull’uso delle preparazioni orali a rilascio modificato di idrocortisone nei pazienti con emoglobinopatia e iposurrenalismo. In caso di eventi acuti, si raccomanda di aumentare il dosaggio della terapia in misura adeguata alla gravità della situazione, con ricorso all’idrocortisone per via parenterale nei casi gravi o di deficit dell’assorbimento (gastro-enterite), oppure per la copertura in occasione di interventi chirurgici o parto. Si raccomanda di istruire attentamente il paziente alla gestione di eventi acuti o intercorrenti e di fornirgli una tessera che documenti la condizione di iposurrenalismo e che contenga le informazioni terapeutiche essenziali.
Nei pazienti con iposurrenalismo in terapia sostitutiva si raccomanda di eseguire monitoraggio clinico e dosaggio di sodio e potassio almeno ogni 6 mesi. La valutazione deve essere particolarmente attenta a una precisa individualizzazione della terapia, al fine di evitare un pericoloso sovra-trattamento che può avere importanti ripercussioni, specie a carattere metabolico ed osseo. Nelle rare forme di insufficienza surrenalica primitiva, va valutata anche l’associazione di basse dosi di fludrocortisone, per coprire il deficit mineraloattivo.
Deficit di GH nell'adulto (R 90-96)
Sebbene il GHD negli adulti con emoglobinopatia sia un problema emergente, la sua prevalenza non è esattamente nota, essendo molto variabile nelle diverse casistiche. Il sospetto clinico può essere difficile, perché segni e sintomi possono essere comuni alla patologia di base; inoltre, il 50% dei pazienti talassemici presenta bassi valori di IGF-1, a genesi multi-fattoriale.
Si suggerisce di eseguire annualmente lo screening per GHD in tutti i soggetti a partire dall’età di 25 anni, mediante valutazione anamnestica e clinica e dosaggio di IGF-1. Nei pazienti con IGF-1 basso, si raccomanda di eseguire il test di stimolo del GH, con GHRH + Arginina come prima scelta. La diagnosi segue i criteri definiti per il GHD nell’adulto dalla nota 39 AIFA: hanno GHD gli adulti con picco di GH, dopo test GHRH + arginina, < 9 µg/L se BMI < 29.9 kg/m2 o < 4 µg/L se BMI ≥ 30 kg/m2. Nei pazienti con GHD, si raccomanda l’esecuzione di RM ipofisaria e il controllo dei restanti assi ormonali.
Si suggerisce di iniziare il trattamento sostitutivo con GH biosintetico solo quando il GHD sia in un appropriato contesto clinico, dimostrato secondo i parametri indicati e in assenza di controindicazioni. Si raccomanda di non iniziare la terapia con GH in presenza di patologia neoplastica maligna in fase di attività e di retinopatia diabetica proliferativa. Si raccomanda di iniziare con dosi di GH biosintetico di 0.2–0.4 mg/die (0.2 mg/die nei maschi e 0.3 mg/die nelle femmine in età pre-climaterica), da somministrare la sera e modulare in base alla risposta clinica, agli eventuali effetti collaterali e ai livelli di IGF-1.
Nei pazienti con GHD in terapia sostitutiva, si raccomanda di eseguire monitoraggio clinico (valutazione BMI, pressione arteriosa, circonferenza addominale, effetti collaterali), metabolico (glicemia, assetto lipidico) e dosaggio di IGF-1 ogni 2 mesi nella fase iniziale, poi ogni 6 mesi. Nei pazienti con GHD in terapia sostitutiva si raccomanda di eseguire: 1) rivalutazione funzionale dell'asse ipofisi-surrene ed eventuale adeguamento della terapia sostitutiva con glucocorticoidi nei pazienti iposurrenalici; 2) valutazione della funzione tiroidea dopo 6 mesi dall’inizio o dalla variazione della terapia con GH.
Età adulta avanzata
La durata di vita del paziente con talassemia major è aumentata nel tempo grazie al miglioramento delle terapie specifiche, e l’invecchiamento rappresenta una nuova frontiera per il paziente talassemico. I pazienti devono continuare a essere monitorati per le complicanze endocrine, sia per diagnosticare quelle di nuova insorgenza, sia per modulare in base all’età ed alle condizioni cliniche le terapie ormonali sostitutive già in atto, tenendo anche conto degli altri farmaci eventualmente assunti per le comorbilità e della loro possibile influenza sugli aspetti endocrini.
Emoglobinopatie non trasfusione-dipendenti
Nelle talassemie non-trasfusione dipendenti e nell’anemia a cellule falciformi le complicanze endocrine sono meno frequenti rispetto ai pazienti trasfusione-dipendenti, ma comunque la loro prevalenza è maggiore rispetto alla popolazione generale, con ulteriore incremento legato all’avanzare dell’età. Pertanto, anche in questi soggetti è richiesto il monitoraggio per la comparsa di endocrinopatie.
Si prevede un aggiornamento periodico delle raccomandazioni in base alle nuove evidenze che si renderanno disponibili. La collaborazione tra specialisti in emoglobinopatie ed endocrinologi dedicati permetterà di migliorare il trattamento delle endocrinopatie nelle emoglobinopatie, accompagnando nelle varie fasi della vita i pazienti, che rivestono un ruolo sempre più consapevole e attivo.
Bibliografia
- Casale M, Baldini MI, Del Monte P, et al. Good clinical practice of the Italian Society of Thalassemia and Haemoglobinopathies (SITE) for the management of endocrine complications in patients with haemoglobinopathies. J Clin Med 2022, 11: 1826.
- Casale M, Baldini MI, Del Monte Pet al. Le principali complicanze endocrinologiche nelle emoglobinopatie: buone pratiche cliniche della Società Italiana Talassemie ed Emoglobinopatie (SITE). Recenti Prog Med 2022, 113: 495-554.
Forme acquisite di tesaurismosi ed endocrinopatie
Marialberta Battocchio, Chiara Martini
Clinica Medica 3°, AO di Padova
AMILOIDOSI
Il termine amiloidosi raggruppa uno spettro di malattie, caratterizzate dalla deposizione progressiva a livello extra-cellulare di amiloide, materiale di derivazione proteica insolubile. Tutte le fibrille di amiloide possiedono un'identica struttura secondaria, con conformazione a foglietto ripiegato ß, e si legano a componenti non fibrillari, quali l'amiloide sierica P (SAP), che hanno una funzione stabilizzatrice. Con la colorazione al rosso Congo l’amiloide presenta una birifrangenza verde mela al microscopio a luce polarizzata. Attualmente si conoscono almeno 30 proteine in grado di generare amiloide nell’uomo.
L’amiloidosi si suddivide in forme localizzate, con deposizione della proteina amiloidogenica nel sito di produzione, limitata quindi a un solo tessuto e in genere associata a processi d’invecchiamento, e in forme sistemiche, in cui la deposizione di amiloide è diffusa a più organi.
| Amiloidosi | ||
| Localizzate | M. di Alzheimer (deposizione SNC) Diabete mellito tipo 2 (deposizione insule pancreatiche) |
|
| Sistemiche | Ereditarie | Deposizione di vari tipi di proteine (transtiretina, lisozima, apolipoproteina A-I e A-II) |
| Acquisite | AL, da catene leggere, associata a differenti forme di discrasia monoclonale delle cellule B AA, complicanza di malattie infiammatorie croniche (TBC, osteomielite cronica, bronchiectasie), connettiviti e neoplasie Nei soggetti dializzati, dovuta alla ridotta escrezione di β2-microglobulina |
|
Nelle amiloidosi sistemiche gli organi più frequentemente coinvolti sono fegato, reni, milza, cuore e sistema gastroenterico, in proporzione differente nelle diverse forme; il coinvolgimento di ghiandole endocrine è descritto, seppur meno frequentemente, ma non sempre al processo infiltrativo consegue un’alterazione funzionale. Le ghiandole più frequentemente colpite sono la tiroide, con il cosiddetto gozzo amiloide, le gonadi maschili e il surrene, mentre più raramente è stato descritto il coinvolgimento di ipofisi, paratiroidi, pancreas e ovaio. La portata epidemiologica del coinvolgimento del sistema endocrino è di difficile valutazione, perché in letteratura sono presenti per lo più case-report. Tuttavia, nella valutazione clinica dell’amiloidosi non è superfluo definire l'interessamento del sistema endocrino, per le possibili complicanze anche gravi legate a quadri disfunzionali.
Surrene nell'amiloidosi sistemica
Il deposito di amiloide a livello surrenalico è stato documentato istologicamente in corso di AL e AA, tuttavia raramente è stata descritta una franca insufficienza surrenalica, essendo necessari un massivo accumulo di amiloide e la conseguente distruzione della corteccia surrenalica perché l’insufficienza funzionale sia conclamata. Più frequentemente si riscontrano livelli di cortisolo nel range basso della norma e una sua ridotta risposta alla stimolazione con ACTH, suggerendo una riduzione della riserva surrenalica. Alla luce di ciò, nei pazienti affetti da amiloidosi sistemica dovrebbero essere ricercate attentamente le manifestazioni cliniche suggestive di insufficienza surrenalica; viene inoltre suggerita l’esecuzione dell’ACTH test.
Gonadi nell'amiloidosi sistemica
Il deposito di amiloide a livello ovarico costituisce un evento raramente descritto. Nelle forme di AA e AL, invece, il coinvolgimento testicolare non è infrequente ed è caratterizzato da abbondante deposito di amiloide a livello dell’epitelio dei tubuli seminiferi, dell'interstizio e del sistema vascolare, con quadri seminali di oligo-azoospermia e funzionali di ipogonadismo ipergonadotropo. Si ipotizza che l’alterazione della spermatogenesi indotta dalla deposizione di amiloide si realizzi attraverso differenti meccanismi (effetto diretto, obliterazione dei canalicoli intra-testicolari e ipossia dovuta al coinvolgimento vascolare, con distruzione dell'epitelio germinale e arresto della spermatogenesi).
Tiroide nell'amiloidosi sistemica
Nel 30-80% dei pazienti con amiloidosi sistemica è descritta una deposizione intra-tiroidea di amiloide, che decorre in maniera asintomatica, mentre è raro il gozzo amiloideo, caratterizzato dall'infiltrazione ghiandolare di amiloide in quantità tale da produrre un ingrandimento clinicamente significativo e per lo più rapido della ghiandola. La sintomatologia, solitamente secondaria all’effetto compressivo che la ghiandola, in rapido aumento dimensionale, esercita sulle strutture adiacenti, è rappresentata da disfagia, dispnea e raucedine. Il gozzo è solitamente di consistenza aumentata e può associarsi a linfadenopatia da deposizione di amiloide. Anche se un ingrandimento bilaterale diffuso, in paziente con nota amiloidosi, è suggestivo più di gozzo amiloideo che di neoplasia, la diagnosi differenziale si impone e per lo più è possibile mediante FNAB. Bisogna comunque ricordare che può verificarsi una deposizione di amiloide nelle aree ghiandolari adiacenti i carcinomi midollari della tiroide.
Istologicamente, oltre alla deposizione parenchimale di amiloide, si rinviene un’infiltrazione adiposa per differenziazione adiposa dei fibroblasti stromali, verosimilmente stimolata dall'ipossia e dalla distruzione dei follicoli tiroidei. L’alterazione funzionale è descritta in circa il 30% dei pazienti con amiloidosi, ma è dovuta solo in parte alla deposizione di amiloide, potendo coesistere tireopatie su base autoimmune e anomalie funzionali tiroidee secondarie all'insufficienza renale cronica, spesso associata a questo tipo di patologie. Il reperto ecografico, non patognomonico, è caratterizzato dall’incremento volumetrico della ghiandola associato a iperecogenicità, ma possono essere presenti anche multiple lesioni solide ipo- o iperecogene e lesioni cistiche.
Altre ghiandole
Nei pazienti anziani (> 80 anni) è stato osservato frequentemente (anche nell’80%) un deposito di amiloide a livello ipofisario; tuttavia, non è ben definito il coinvolgimento di tale ghiandola nelle forme sistemiche. Infatti, la barriera emato-encefalica non permette il passaggio delle fibrille di amiloide e solitamente la funzione ipofisaria è conservata nelle forme di amiloidosi sistemica, sebbene in letteratura sia descritto qualche caso di ipopituitarismo.
Nei pazienti con amiloidosi sistemica è stato descritto un interessamento delle paratiroidi, ma è raro che tale coinvolgimento determini un'alterazione funzionale.
Più frequente nelle forme sistemiche è l'interessamento del pancreas, ma ancora una volta la sua compromissione funzionale è ben più rara.
EMOCROMATOSI
L'emocromatosi, intesa come condizione caratterizzata da sovraccarico di ferro, racchiude un ampio spettro di malattie: forme ereditarie, forme secondarie ad altre patologie, forme secondarie al trattamento trasfusionale per patologie ematologiche.
L’emocromatosi ereditaria (HH) è causata da differenti mutazioni geniche che interessano la funzione di proteine implicate nell’assorbimento e nell’immagazzinamento del Fe, determinandone un sovraccarico. L’accumulo di ferro tende a localizzarsi a livello delle cellule parenchimali, danneggiandole attraverso la produzione di radicali liberi dell’ossigeno. Perché il sovraccarico divenga clinicamente evidente, è tuttavia importante la presenza di fattori ereditari e acquisiti favorenti, come sesso, consumo di alcol o presenza di sindrome metabolica; pertanto, l’espressione fenotipica è molto variabile e in genere compare in età adulta se si esclude la rara forma di emocromatosi giovanile (JH). Da quando è disponibile l'analisi genetica e sono maggiormente noti i meccanismi patogenetici delle forme ereditarie, le manifestazioni cliniche conclamate sono sempre più rare.
Nelle forme di emocromatosi secondaria, quali quelle che accompagnano malattie diseritropietiche come la talassemia major, la talassemia intermedia e l’anemia aplastica, il sovraccarico di ferro tende inizialmente a localizzarsi a livello delle cellule del sistema reticoloendoteliale, e deriva da processi di emolisi, aumento dell’assorbimento di ferro dovuto a eritropoiesi inefficace o sovraccarico secondario alla terapia trasfusionale cronica. Le manifestazioni cliniche, nonostante la terapia chelante, possono comparire precocemente, in quanto le trasfusioni vengono utilizzate fin dai primi mesi di vita.
Gli organi/sistemi maggiormente colpiti dall’accumulo di ferro sono fegato, cuore e sistema endocrino.
Pancreas
Il diabete è da sempre considerata una delle manifestazioni cliniche dell'emocromatosi, sia delle forme ereditarie, che di quelle secondarie. I suoi momenti patogenetici sono il danno β-cellulare e l'insulino-resistenza legata al danno epatico. Il sovraccarico di ferro sottopone le β-cellule a stress ossidativo, provocando ridotta sensibilità allo stimolo glucidico, conseguente deficit di secrezione insulinica oltre che apoptosi cellulare. L'insulino-resistenza sembra un evento tardivo nella transizione tra la ridotta tolleranza glucidica e diabete franco e solitamente è correlata all'obesità e/o al danno epatico ferro-indotto. La ridotta tolleranza glucidica e il diabete sono manifestazioni annoverate tra le complicanze della terapia trasfusionale cronica, manifestandosi quando concomita una compromissione epatica.
Funzione gonadica
Nelle forme di sovraccarico di ferro l'ipogonadismo è solitamente secondario, dovuto cioè a un danno ipofisario, ove il Fe danneggia preferenzialmente le cellule gonadotrope, determinando una ridotta secrezione di gonadotropine. Le indagini ormonali in genere documentano livelli bassi di gonadotropine basali o inappropriatamente normali in relazione a quelli di testosterone o 17β-estradiolo, e una loro ridotta o assente risposta allo stimolo con GnRH. Sono state descritte anche forme di ipogonadismo primario, solitamente in malattie in stadio avanzato e con sovraccarico di ferro di grado marcato. Nell'emocromatosi ereditaria la prevalenza dell'ipogonadismo dipende dal tipo, e in genere si manifesta verso la fine della seconda decade di vita. Quando il sovraccarico di ferro è secondario a terapia trasfusionale, l’ipogonadismo è relativamente frequente e direttamente correlato ai livelli di ferritina. Nelle talassemie, inoltre, il danno ferro-mediato, se non adeguatamente prevenuto dalla terapia chelante, può produrre ritardo o arresto dello sviluppo puberale.
Nei soggetti talassemici è quindi indicata una valutazione clinica e dello stadio di Tanner ogni 6 mesi prima della pubertà e l'esecuzione annuale di esami bioumorali, oltre che morfologici per indagare l’eventuale ritardo puberale.
Ipofisi
Come già detto, il danno da accumulo di Fe a livello ipofisario sembra prevalentemente a carico delle cellule gonadotrope e il panipopituitarismo è un evento raro in questo tipo di condizione. Nell’ambito dell’emocromatosi ereditaria sono state comunque descritte una ridotta risposta di PRL allo stimolo con TRH e un’alterata risposta del GH allo stimolo ipoglicemico e con GHRH, ma la presenza di un ipogonadismo non corretto potrebbe condizionare la risposta del GH agli stimoli. Anche nel setting dei pazienti in terapia trasfusionale cronica è stata ugualmente descritta una ridotta risposta della PRL agli stimoli, mentre più rara appare un’alterazione della secrezione di GH. Infine, nei soggetti talassemici il ritardo di crescita secondario a deficit di GH è un evento frequente e correlato non solo al sovraccarico di ferro, ma anche a deficit nutrizionali e all’uso intensivo di terapia chelante con desferoxamina. Inoltre in questi pazienti viene suggerita un’alterazione della regolazione ipotalamica della secrezione di GH. Per questo ancora una volta nei soggetti talassemici vanno attentamente monitorati crescita, età ossea e assetto ormonale (test di stimolo per GH, IGF-1 e IGF-BP3).
Tiroide
Sebbene il ferro tenda a depositarsi anche nel tessuto tiroideo, l’alterazione funzionale è stata descritta raramente e solitamente in soggetti con cirrosi epatica e/o altri disordini endocrini. Una disfunzione tiroidea primitiva appare più una coincidenza che una vera associazione nell’ambito delle emocromatosi ereditarie. Nei pazienti talassemici l’ipotiroidismo rappresenta invece una conseguenza tardiva dell’accumulo di Fe nella ghiandola, con conseguente fibrosi. Splenectomia, scarsa compliance alla terapia chelante e alti livelli di ferritina sono fattori favorenti il suo sviluppo.
Surrene
Il ferro si deposita principalmente nella zona glomerulosa della ghiandola surrenalica, ma questo raramente comporta un deficit di secrezione di aldosterone, anche nei soggetti affetti da emocromatosi ereditaria. La secrezione di glucocorticoidi è in genere conservata. Nei soggetti talassemici non deve essere misconosciuta la possibilità di una ridotta riserva surrenalica, condizione che può aggravare la mortalità in corso di eventi acuti come lo scompenso cardiaco o la sepsi. Non è ancora chiaro a tutt’oggi con quale meccanismo si realizzi il deficit funzionale ed è stata anche suggerita un’alterata regolazione ipotalamica, al pari di quanto avviene per la secrezione di GH.
Paratiroidi e osso
Sebbene l’accumulo di Fe nelle paratiroidi sia ampiamente descritto, le alterazioni funzionali sono estremamente rare nei pazienti affetti da emocromatosi ereditaria, più frequenti nei pazienti talassemici, per cui è fondamentale la terapia chelante in prevenzione primaria ed eventualmente il trattamento con calcitriolo.
L’osteoporosi è una frequente manifestazione del sovraccarico di ferro, correlata alla sua gravità, indipendentemente dalla presenza di ipogonadismo, cirrosi e dai livelli di vitamina D, tutti fattori influenzanti la BMD. L’eccesso di Fe esercita una tossicità diretta sugli osteoblasti, inibendone la proliferazione e l’attività di osteoformazione. L’eventuale concomitanza di ipogonadismo concorre all’evoluzione del quadro, stimolando il riassorbimento osseo. Dal momento che i diversi background genetici dell’emocromatosi ereditaria non determinano variazioni di prevalenza dell’osteoporosi, sembra che la responsabilità diretta sia da attribuire al sovraccarico di ferro. Questa interpretazione è supportata dall’alta prevalenza di osteoporosi e osteopenia nelle condizioni di sovraccarico di ferro dovute a terapia trasfusionale cronica, in cui la gravità è correlata ai livelli di ferritina. Nelle talassemie, inoltre, altri fattori concorrono all’alterazione del metabolismo osseo, come l’espansione del midollo osseo per eritropoiesi inefficace, il deficit di vitamina D, fattori genetici, il sesso e la ridotta attività fisica.
BIBLIOGRAFIA
- Ozdemir D, Dagdelen S, Erbas T. Endocrine involvement in systemic amyloidosis. Endocr Pract 2010, 16: 1056-63.
- Ozdemir D, Dagdelen S, Erbas T, et al. Amyloid goiter and hypopituitarism in patient with systemic amyloidosis. Amyloid 2011, 18: 32-4.
- Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet 2016, 387: 2641–54.
- Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet 2016, 388: 706–16.
- Pelusi C, Gasparini DI, Bianchi N, Pasquali R. Endocrine dysfunction in hereditary hemochromatosis. J Endocrinol Invest 2016, 39: 837–47.
- Kim MK, Lee JW, Baek KH, et al. Endocrinopathies in transfusion-associated iron overload. Clin Endocrinol 2013, 78: 271-7.
- Inati A, Noureldine MA, Mansour A, Abbas HA. Endocrine and bone complications in β-thalassemia intermedia: current understanding and treatment. Biomed Res Int 2015, 2015: 813098.
Anemie ed endocrinopatie
Ilaria Tenuti
Medicina Generale, AOU Verona
(aggiornato al maggio 2024)
PREMESSA
È noto come spesso le endocrinopatie siano accompagnate da quadri di anemia di vario tipo ed è altresì noto che le anemie spesso possano sottendere un'endocrinopatia misconosciuta. Il rapporto tra sistema endocrino ed anemia è quindi biunivoco (1).
Molti studi hanno evidenziato come gli ormoni intervengano sull'eritropoiesi attraverso meccanismi diversi:
- stimolando la produzione renale di eritropoietina (EPO);
- favorendo l'attività dell'EPO sui precursori midollari;
- stimolando direttamente il midollo stesso.
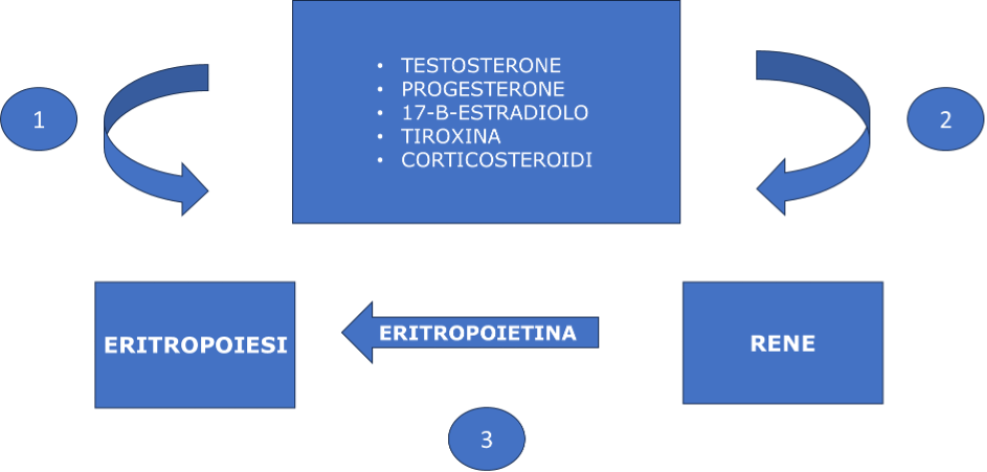
Si deve inoltre ricordare che i processi immunitari, spesso associati alle endocrinopatie, e alcuni farmaci impiegati nella terapia possono interferire con l’assorbimento dei componenti fondamentali per l’eritropoiesi e con l’emivita dei globuli rossi.
Le alterazioni ormonali alla base di una singola endocrinopatia possono essere associate a più forme di anemia, che hanno un ruolo importante nella progressione delle complicanze.
DISFUNZIONI TIROIDEE
Anemia e disfunzione tiroidea si verificano spesso contemporaneamente, ma rimane poco chiaro il comune denominatore dei due disordini (2). Sappiamo che gli ormoni tiroidei stimolano la proliferazione degli eritrociti (3):
- direttamente, potenziando l'effetto dell'EPO su colonie eritroidi;
- indirettamente, aumentando la produzione di EPO in risposta all'ipossia.
Ipotiroidismo
L’anemia, che in molti pazienti può essere il primo segno di ipotiroidismo, ha fenotipi molto variabili: normocitica, microcitica o macrocitica. L’anemia che si osserva nei pazienti ipotiroidei è anche influenzata da possibili carenze nutrizionali associate: ciò rende più difficile una sua definizione. Come è noto, gli ipotiroidei autoimmuni hanno elevata incidenza di anemia megaloblastica, dovuta alla carenza di fattore intrinseco per presenza di gastrite atrofica.
La forma più frequente è l’anemia da carenza di ferro: se questa è stata causata dall’ipotiroidismo, sarà necessario trattare quest’ultimo in modo adeguato per ottenere valori emocromocitometrici accettabili. L'anemia sideropenica:
- influenza negativamente lo stato tiroideo, poiché il ferro è vitale per l'attività della perossidasi tiroidea, l’enzima contenente ferro, fondamentale nelle prime tappe della sintesi dell'ormone tiroideo;
- può indurre alterazioni nel controllo del sistema nervoso centrale sull'asse tiroideo e ridurre il legame della T3 ai suoi recettori epatici;
- compromette il metabolismo della tiroide, anche diminuendo il trasporto di ossigeno.
È importante ricordare che il livello di ferro nel nostro organismo può essere in grado di modificare la risposta alla profilassi con lo iodio, che infatti risulta meno efficace nei soggetti con anemia e/o nei soggetti con risposta più scarsa al trattamento marziale.
Ipertiroidismo
Il meccanismo con cui si sviluppa l’anemia nel paziente affetto da ipertiroidismo è meno chiaro. Spesso negli ipertiroidei sono stati rilevati elevati livelli di EPO. Un’ipotesi fisiopatologica potrebbe essere un’alterato metabolismo del ferro, che, associato a stress ossidativo, indurrebbe maggiore fragilità degli eritrociti, con conseguente riduzione della loro sopravvivenza (2).
Alla luce di quanto riportato, si consiglia di valutare lo stato ematologico nei pazienti con disturbi tiroidei, poiché l’anemia in pazienti con distiroidismi è condizione frequente, spesso non riconosciuta. È altresì utile nella pratica clinica prendere in considerazione possibili disfunzioni tiroidee sia nella diagnosi differenziale di anemia di ndd, sia in caso di anemia resistente al trattamento.
IPOSURRENALISMO
Studi sperimentali hanno evidenziato come la surrenectomia causi una moderata anemia, responsiva alla terapia con glucocorticoidi o EPO.
Nei pazienti con morbo di Addison si osserva un’anemia normocitica/normocromica, simile a quella del modello animale. L’anemia dell’iposurrenalico viene spesso sottovalutata per la concomitante riduzione del volume plasmatico che altera la concentrazione emoglobinica e l’ematocrito, quindi non correlata alla riduzione del volume eritrocitario.
Il ruolo dei glucocorticoidi nell’anemia non appare chiaro: a dosi fisiologiche hanno solo effetti lievi sull’eritropoiesi, a dosi farmacologiche causano modesta eritrocitosi. Tuttavia, non è stato ancora definito se l’effetto degli steroidi surrenalici sia diretto sui precursori eritroidi o mediato da EPO.
IPOPITUITARISMO
Nei pazienti ipofisectomizzati o con ipopituitarismo si osserva anemia normocromica/normocitica da ridotta produzione midollare. L’anemia è corretta da terapia sostitutiva tiroidea, surrenalica e gonadica.
Modelli sperimentali e dati di letteratura mettono in rilievo il ruolo dell’IGF-1 nella modulazione dell’eritropoiesi. Nella pratica clinica in soggetti affetti da deficit di GH si registra il miglioramento della crasi ematica a seguito della somministrazione di GH, effetto verosimilmente correlato all’aumento dei valori di IGF-1, in assenza di alterazioni dei livelli di EPO (4).
IPERPARATIROIDISMO
Talvolta nell’anemia può essere osservato un quadro di iperparatiroidismo primitivo (5). Per quanto riguarda le possibili relazioni tra iperparatiroidismo e anemia, elevati valori di PTH interferiscono con la normale eritropoiesi attraverso un duplice meccanismo:
- a livello midollare tramite la down-regulation dei recetttori per l’EPO sui progenitori eritroidi, con meccanismo non del tutto chiaro;
- a livello periferico, aumentando la permeabilità degli eritrociti al calcio, favorendone l’ingresso all’interno della cellula, con conseguente incremento della fragilità osmotica e lisi cellulare dell’eritrocita.
L’anemia può essere causata anche da un meccanismo indiretto:
- da danno renale come in caso di nefrocalcinosi;
- da mielosclerosi, con riduzione della proliferazione dei precursori eritroidi.
DISFUNZIONE GONADICHE
L’anemia è tra i sintomi che si manifestano con maggiore frequenza nei soggetti con carenza di testosterone, mentre è ben noto l’effetto positivo degli androgeni sull’eritropoiesi, sia a livello fisiologico che farmacologico, motivo per cui sono stati utilizzati in terapia, indipendentemente dalla causa dell’anemia.
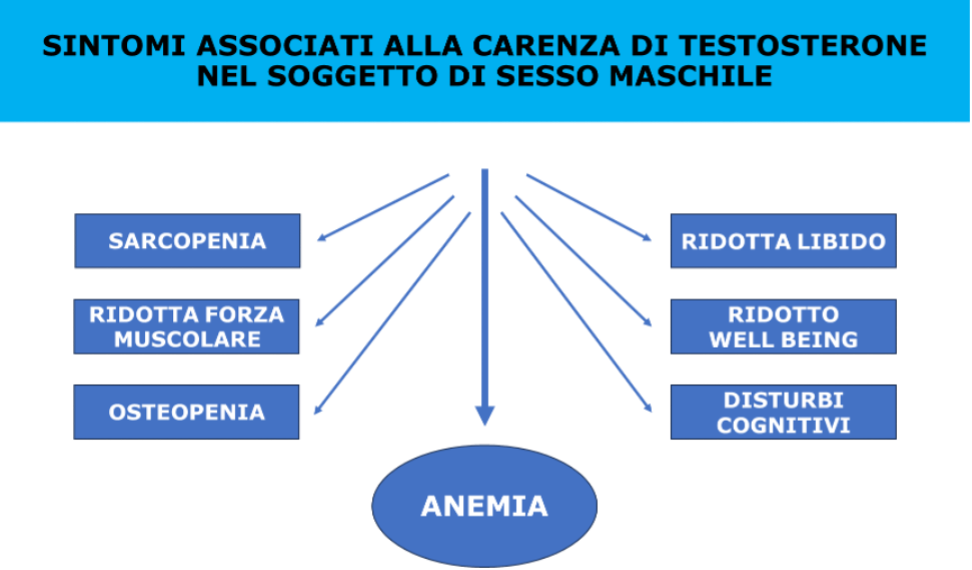
Il meccanismo tramite cui il testosterone agisce sul sistema emopoietico è duplice:
- diretto:
- aumenta l’attività del midollo osseo (stimolazione CFU-E);
- stimola l’incorporazione del ferro nei globuli rossi;
- aumenta la captazione del glucosio con attivazione della glicolisi;
- potenzia l’effetto dell’IGF-1, con maturazione e proliferazione dei pro-eritroblasti;
- aumenta l’emivita dei globuli rossi;
- indiretto:
- stimola l’EPO sul potenziamento dell’attività dell’RNA-polimerasi, con incremento della massa renale (6).
Gli androgeni a dosi farmacologiche stimolano la produzione di globuli rossi attraverso un documentato aumento di EPO. In un gruppo di uomini trattati con testosterone per 36 mesi i livelli di emoglobina risultavano significativamente aumentati rispetto al gruppo trattato con placebo (7). Uno studio del 2018 aveva l’obiettivo di determinare se la somministrazione di testosterone, in uomini di età ≥ 65 anni (età media 74.8 anni, BMI medio 30.7) che presentavano bassi livelli di testosterone e anemia inspiegabile, fosse in grado di incrementare i livelli di emoglobina. Il trattamento con testosterone ha determinato un aumento dei livelli di emoglobina di 1 g/dL rispetto al basale nel 54% dei casi (8). Questo studio indica che negli uomini ultra65enni potrebbe essere utile misurare i livelli di testosterone al fine di valutare la possibilità di utilizzare l’ormone come strumento terapeutico, anche se sono necessari ulteriori studi, dati i noti effetti collaterali del testosterone.
DIABETE MELLITO tipo 2
La coesistenza di anemia col diabete mellito è evidenza clinica rilevante: si calcola che la prevalenza di anemia sia del 14-45% nei soggetti diabetici. In questi, inoltre, il rischio di sviluppare anemia è circa 2-3 volte maggiore che nei non diabetici.
Il rischio di sviluppare anemia è prevalentemente legato al danno renale, ma la patogenesi dell’anemia nel DM2 è multi-fattoriale e complessa:
- carenza di ferro:
- iporesponsività all’EPO, dovuta alla sua glicazione e a quella del suo recettore;
- insufficienza renale;
- neuropatia autonomica con riduzione della stimolazione adrenergica per la produzione di EPO;
- ipogonadismo con ridotti valori di testosterone;
- elevati livelli di citochine pro-infiammatorie, che favoriscono l’apoptosi dei precursori eritroidi;
- proteinuria con eliminazione di EPO con le urine.
Sempre in pazienti con DM2 la terapia con ipoglicemizzanti orali può contribuire all’insorgenza di anemia, in quanto la metformina riduce l’assorbimento di vitamina B12 dopo 4 mesi di somministrazione. I dati di letteratura riportano che l’anemia megaloblastica può insorgere anche dopo 5-10 anni di terapia con metformina, a causa delle riserve epatiche di vitamina B12. Quindi appare utile dosare periodicamente i livelli di vitamina B12 nei pazienti che assumono metformina. I tiazolidinedioni possono contribuire all’anemia, favorendo l’accumulo midollare di grasso con riduzione della massa eritroide. Al contrario, gli inibitori del DPP-4 migliorano l’anemia, aumentando i livelli di EPO, data la riduzione della sua degradazione da parte del DPP-4.
L’anemia del diabetico, causata in parte dall’insufficienza renale, può a sua volta indurre la progressione della nefropatia diabetica, favorendo un circolo vizioso che porta all’insufficienza renale terminale, con aumento della pressione glomerurale e proteinuria. La presenza di anemia nel diabetico incrementa il rischio di progressione delle complicanze micro- e macro-vascolari ed è un fattore di rischio indipendente di mortalità cardio-vascolare (associandosi a ipertrofia del ventricolo sinistro e scompenso cardiaco). Da cui si evince come la correzione dell’anemia nel diabetico riduca l’insorgenza di complicanze e di mortalità (9).
UN LINK AL CONTRARIO: TALASSEMIA
Negli ultimi decenni, i soggetti talassemici sopravvivono a lungo in relazione al miglioramento delle cure, ma vanno incontro a malattie da accumulo di ferro nei parenchimi ghiandolari, con conseguenti ipofunzioni endocrine:
- ipogonadismo ipogonadotropo per deficit di LH ed FSH (50% dei casi) (10);
- deficit di GH, che si manifesta come ritardo nella crescita (15-20%);
- IGT e DM2 (10%).
CONCLUSIONI
- L’anemia può essere conseguenza dell’endocrinopatia e va trattata se persiste anche dopo la correzione dei valori ormonali.
- Si devono valutare le concause dell’anemia, che eventualmente vanno corrette (deficit di vitamina B12, folati, ferro).
- Nel diabetico l’anemia deve far parte della valutazione delle complicanze ed essere corretta in base al meccanismo patogenetico prevalente. Un’attenzione particolare va posta all’effetto sull’anemia della terapia ipoglicemizzante.
- Servono ulteriori studi per definire il ruolo di IGF-1 e degli ormoni tiroidei.
- Un’anemia da cause inspiegabili potrebbe essere spiegata da un’endocrinopatia misconosciuta.
BIBLIOGRAFIA
- Piticchio T, Frasca F. Endocrinopatie e anemie/anemie ed endocrinopatie: una si(pali)ndrome spesso misconosciuta. L’endocrinologo 2020, 21: 277-83.
- Sczepanek-Parulska E, et al. Anemia in thyroid diseases. Pol Arch Intern Med 2017, 127: 352-60.
- Golde DW, et al. Thyroid hormones stimulate erythropoiesis in vitro. Br J Haematol 1977, 37: 173-7.
- Christ ER, et al. The importance of growth hormone in the regulation of erythropoiesis, red cell mass, and plasma volume in adults with growth hormone deficiency. J Clin Endocrinol Metab 1997, 82: 2985-90.
- Boxer M, et al. Anemia in primary hyperparathyroidism. Arch Intern Med 1977, 137: 588-90.
- Shahani S, et al. Androgens and erythropoiesis: past and present. J Endocrinol Invest 2009, 32: 704-16.
- Maggio M, et al. Is the haematopoietic effect of testosterone mediated by erythropoietin? The results of a clinical trial in older men. Andrology 2013, 1: 24-8.
- Roy CN, et al. Association of testosterone levels with anemia in older men. A controlled clinical trial. JAMA Internal Med 2017, 177: 480-90.
- Sahay M, et al. Diabetes and anemia: International Diabetes Federation (IDF)-Southeast Asian region (SEAR) position statement. Diabetes Metab Syndr 2017, 11 suppl 2: S685-95.
- De Sanctis V, et al. Growth and endocrine complications in thalassemia. The international network on endocrine complications in thalassemia (I-CET) position statement and guidelines. Indian J Endocrinol Metab 2013, 17: 8-18.