dell'iperparatiroidismo
Aspetti specifici nella terapia dell'ipoparatiroidismo

Guido Zavatta
Dipartimento di Scienze Mediche e Chirurgiche, Università di Bologna, Alma Mater Studiorum; UOC Endocrinologia, Prevenzione e Cura del Diabete, IRCCS AOU di Bologna, Policlinico S. Orsola
(aggiornato al 30/6/2025)
Strategie di prevenzione dell’ipocalcemia post-operatoria
Le linee guida dell’American Thyroid Association raccomandano di valutare i livelli di 25OH-vitamina D prima della tiroidectomia e di correggere eventuali carenze prima dell’intervento. La carenza di vitamina D è, infatti, associata ad aumentato rischio di ipocalcemia post-operatoria, soprattutto nella forma transitoria. La supplementazione pre-operatoria di vitamina D, eventualmente associata al calcio, può ridurre l’incidenza e la durata dell’ipocalcemia dopo l’intervento (1).
Crisi ipocalcemica
L’ipocalcemia acuta può rappresentare una condizione potenzialmente fatale nei pazienti con ipoparatiroidismo cronico, soprattutto se insorge rapidamente (ad esempio, dopo un intervento di chirurgia anteriore del collo in assenza di adeguata supplementazione di calcio o vitamina D). In tali casi, è raccomandata la somministrazione endovenosa di gluconato di calcio, sotto monitoraggio ECG continuo, poiché una correzione troppo rapida può a sua volta indurre aritmie fino all’arresto cardiaco (2). La dose iniziale raccomandata è generalmente di 1-2 grammi di gluconato di calcio, in pratica 1-2 fiale da 10 mL di calcio gluconato al 10%, diluite in 50 mL di glucosata al 5% o di soluzione fisiologica, da infondere in 10-20 minuti. Successivamente, si consiglia di avviare un'infusione più lenta (0.5-1.5 mg/kg/ora), da proseguire per almeno 8-10 ore. Il gluconato di calcio contiene 93 mg di calcio elementare ogni 10 mL di soluzione alla concentrazione standard del 10%. La calcemia sierica andrebbe monitorata ogni 4-6 ore. Nei pazienti che presentano anche acidosi è fondamentale correggere prioritariamente l’ipocalcemia prima di intervenire sul disturbo acido-base, poiché una correzione inversa ridurrebbe ulteriormente la quota di calcio ionizzato (3).
A differenza dell’uso dell’insulina nelle crisi iperglicemiche, gli analoghi del PTH somministrati per via sottocutanea non sono indicati per il trattamento dell’ipocalcemia acuta da ipoparatiroidismo, poiché questa via di somministrazione potrebbe in realtà ritardare l’effetto clinico del farmaco. Tuttavia, è interessante notare che il PTH 1-34 sembra in grado di prevenire l’ipocalcemia acuta post-tiroidectomia se somministrato 4 ore dopo l’intervento e successivamente ogni 12 ore fino alla dimissione. Nello studio THYPOS, i pazienti con PTH intatto < 10 pg/mL a 4 ore dall’intervento erano stati randomizzati a ricevere PTH 1-34 oppure terapia standard: il gruppo trattato con PTH 1-34 mostrava calcemia più alta alla dimissione (8.5 vs 7.8 mg/dL) e degenza ospedaliera più breve (2 vs 3 giorni) (4).
Inquadramento dell’ipoparatiroidismo nelle prime giornate post-operatorie
Secondo le ultime linee guida del II International Workshop, la misurazione del PTH sierico poche ore dopo la tiroidectomia totale può essere utilizzata per prevedere quali pazienti non svilupperanno ipoparatiroidismo post-chirurgico permanente (raccomandazione forte, evidenza di qualità moderata) (5): se i valori di PTH sono > 10 pg/mL (1.05 pmol/L) a 12–24 ore dall’intervento, è improbabile che il paziente sviluppi ipoparatiroidismo permanente; pertanto, non sarà necessario un trattamento a lungo termine con vitamina D attiva e supplementi di calcio. Molti pazienti con valori di PTH < 10 pg/mL (1.05 pmol/L) nelle prime 12–24 ore dopo l’intervento possono comunque andare incontro al recupero di un ipoparatiroidismo transitorio. L’ipoparatiroidismo transitorio è, infatti, una condizione frequente dopo tiroidectomia totale e può risolversi spontaneamente nel tempo. Il monitoraggio clinico e biochimico deve pertanto proseguire nei mesi successivi all’intervento chirurgico, per distinguere le forme transitorie da quelle permanenti, che vengono definite tali solo se persistenti oltre 12 mesi (6).
Gestione dell’immediato post-operatorio e nelle prime settimane
Nei primi giorni successivi alla tiroidectomia totale la gestione dell’ipoparatiroidismo post-operatorio richiede monitoraggio ravvicinato e interventi terapeutici tempestivi per prevenire l’ipocalcemia sintomatica. In questi casi, si raccomanda l’inizio precoce della supplementazione orale con calcio a dosaggi di 1–2 g/die, suddivisi in 2–3 somministrazioni. Nei quadri più lievi si può iniziare da 0.5–1 g/die, mentre in situazioni più gravi o con ipocalcemia persistente si può salire progressivamente fino a 3–4 g/die, sotto stretto controllo clinico e laboratoristico. In parallelo, si inizia calcitriolo alla dose iniziale di 0.25–0.5 µg/die, con possibilità di aumento fino a 2.0 µg/die in base alla risposta clinica o ai fattori di rischio pre-esistenti all’intervento chirurgico (es. rischio di hungry bone syndrome). In caso di ipocalcemia sintomatica (calcemia < 7.5 mg/dL e/o segni/sintomi neuromuscolari), è indicata la somministrazione endovenosa di calcio gluconato, seguita eventualmente da infusione continua. Nella prima settimana va eseguito il monitoraggio ogni 24–48 ore dei livelli ematici di calcio (ionizzato o corretto per albumina), fosfato, magnesio e creatinina. L’ipomagnesiemia deve essere corretta, perché può ostacolare l’efficacia del trattamento dell’ipocalcemia. Alla dimissione, la terapia va personalizzata sulla base dei valori più recenti, con follow-up clinico e biochimico a 7–14 giorni. Le prime settimane sono critiche per la stabilizzazione della calcemia e richiedono un contatto clinico ravvicinato. Dopo 2–4 settimane, le linee guida ATA suggeriscono di eseguire dosaggio della calciuria delle 24 ore per escludere ipercalciuria (> 250–300 mg/die), che richiederebbe nuovi aggiustamenti posologici. L’obiettivo terapeutico resta il mantenimento della calcemia al limite basso del range normale, evitando precipitazione dei sintomi o sovra-trattamento (1).
Supplementi di calcio nella terapia dell’ipoparatiroidismo: novità sul trattamento ‘convenzionale’?
La supplementazione orale di calcio può essere effettuata sotto forma di carbonato di calcio, che contiene circa il 40% di calcio elementare, oppure di citrato di calcio, che ne contiene circa il 21%. Per il trattamento dell’ipoparatiroidismo cronico non sono raccomandate, per il basso contenuto di calcio elementare, altre formulazioni, come il lattato (13% di calcio elementare) e il gluconato (9%). È importante ricordare che il calcio orale agisce anche come chelante del fosforo, risultando utile nel ridurre l’iperfosfatemia. Si raccomanda che il carbonato di calcio venga assunto durante i pasti, poiché l’assorbimento richiede l’acidità del pH gastrico. Il citrato di calcio, invece, non necessita di acidità gastrica per l’assorbimento e può essere assunto anche a stomaco vuoto (7).
Alcune nuove evidenze sul trattamento convenzionale dell’ipoparatiroidismo provengono da uno studio randomizzato, in doppio cieco, cross-over, condotto su 24 pazienti con ipoparatiroidismo post-chirurgico, che ha confrontato citrato di calcio e carbonato di calcio (8): il citrato è risultato meglio tollerato (con minore incidenza di stipsi) e ha determinato riduzione significativa del rapporto ossalato/creatinina. Questi risultati possono avere implicazioni cliniche nei pazienti con nefrolitiasi o con disturbi gastro-intestinali imputabili alla supplementazione di calcio.
Ipercalciuria e diuretici tiazidici
Nei pazienti con ipercalciuria persistente può essere utile l’introduzione di diuretici tiazidici in associazione a una dieta povera di sodio, con attento monitoraggio dei livelli sierici di potassio e magnesio. Le evidenze a supporto dell’uso dei tiazidici nei pazienti con ipoparatiroidismo derivano da un piccolo studio pilota (9). Tuttavia, uno studio longitudinale retrospettivo su pazienti con ipoparatiroidismo permanente ha mostrato che pazienti in trattamento con tiazidici presentavano livelli di calciuria più elevati rispetto a quelli non trattati (10). Considerando che le evidenze relative all’efficacia dei tiazidici sono contrastanti, è necessario ricorrere al giudizio clinico per valutarne l’impiego caso per caso. Nei pazienti trattati con tiazidici è raccomandato un attento monitoraggio della pressione arteriosa, della funzione renale e dei livelli sierici di magnesio e potassio. Inoltre, è opportuno informare i pazienti che l’uso di questi farmaci è stato associato ad aumentato rischio di alcuni tumori cutanei (11,12), pertanto devono controllare regolarmente la cute per identificare eventuali nuove lesioni o modifiche di quelle esistenti e a segnalare al medico ogni lesione cutanea sospetta (13). I tiazidici vanno evitati nei pazienti con ipocalcemia autosomica dominante di tipo 1 o 2, poiché possono peggiorare l’ipomagnesiemia (14). Sono inoltre controindicati nei pazienti con insufficienza surrenalica, manifestazione chiave della sindrome polighiandolare autoimmune di tipo 1 (APS-1).
Ipomagnesiemia
L’ipomagnesiemia può determinare resistenza all’azione del PTH e alterare pertanto la risposta alla terapia convenzionale. È quindi fondamentale correggere la causa sottostante dell’ipomagnesiemia. In caso di ipomagnesiemia grave (< 1 mg/dL) e sintomatica è necessaria la somministrazione endovenosa di magnesio. Nei casi asintomatici è invece possibile correggere il deficit con una terapia orale. Le preparazioni orali dovrebbero sempre essere iniziate a dosaggi minimi, con incremento graduale, al fine di minimizzare l’insorgenza di diarrea, che rappresenta l’effetto avverso principale della supplementazione di magnesio (1).
Quando valutare la terapia con analoghi del paratormone
Le linee guida internazionali riportate in tabella 1 sono state redatte prima dell'approvazione da parte di EMA e FDA di palopegteriparatide come trattamento per l’ipoparatiroidismo; pertanto, si basano essenzialmente sui numerosi studi riguardanti PTH(1-84), farmaco differente da palopegteriparatide (15). Infatti, il PTH(1-84) era stato approvato come terapia aggiuntiva o ‘add-on’ alla terapia convenzionale in pazienti con ipoparatiroidismo, al contrario di palopegteriparatide che ha ricevuto la registrazione da EMA come terapia sostitutiva per l’ipoparatiroidismo cronico.
| Tabella 1 Indicazioni per la terapia a base di analoghi del paratormone |
|
| II Workshop Internazionale (2022) (16,17) |
La terapia con PTH dovrebbe essere presa in considerazione nei pazienti:
La terapia con PTH può essere considerata in soggetti con osteoporosi che richiedano trattamento anti-riassorbitivo (es. denosumab), per ridurre il rischio di ipocalcemia. |
| PARAT Consensus Europea (2022) (17) | Nessuna raccomandazione specifica fornita. “Il PTH(1-34), somministrato una o due volte al giorno, potrebbe rappresentare un'opzione alternativa, sebbene con meno dati di sicurezza” |
Palopegteriparatide (TransCon-PTH)
Quasi contestualmente al ritiro definitivo dal commercio di PTH(1-84) (Natpara®) nel dicembre 2024, palopegteriparatide ha ricevuto l’autorizzazione da EMA (11/2023) e FDA (08/2024) ed è ora approvata come prima terapia sostitutiva per l’ipoparatiroidismo cronico, anche se non è ancora disponibile in tutti gli stati europei.
Palopegteriparatide, noto precedentemente anche come TransConPTH(1-34), è un pro-farmaco costituito da teriparatide coniugato a un carrier di metossi-polietilen-glicole tramite un linker, che consente il rilascio prolungato del principio attivo nell’arco di 24 ore (tecnologia TransCon). L’emivita del farmaco raggiunge le 60 ore, grazie al rilascio graduale (dipendente da pH e temperatura corporea) del teriparatide nel circolo sanguigno. Questo meccanismo consente la stimolazione continuativa del recettore del PTH, come se fosse in corso un’infusione continua, mimando pertanto la continuità di azione dell’ormone endogeno sul proprio recettore.
Nello studio di fase 2 PaTH Forward, TransCon PTH ha consentito al 91% dei pazienti di sospendere la vitamina D attiva e i supplementi di calcio (intesi come > 500 mg/die), con miglioramento della qualità di vita e normalizzazione dei parametri biochimici (18).
Lo studio multicentrico di fase 3 PaTHway, mirato a valutare efficacia e sicurezza di palopegteriparatide, era strutturato in due fasi: una fase iniziale in doppio cieco di 26 settimane con randomizzazione a farmaco o placebo, seguita da una fase in aperto di 156 settimane, durante la quale tutti i partecipanti hanno ricevuto il trattamento con palopegteriparatide (19). La popolazione iniziale comprendeva 82 adulti con ipoparatiroidismo cronico da almeno 6 mesi, con diverse eziologie (prevalentemente post-chirurgica), di età media 48.6 anni e durata media della malattia di 11.7 anni. L’end-point primario era il raggiungimento della calcemia corretta nel range di normalità (8.3–10.6 mg/dL) con indipendenza da calcitriolo e supplementazione di calcio > 600 mg/die. Alla 26° settimana questo obiettivo era stato raggiunto nel 79% dei pazienti nel braccio attivo contro il 5% del gruppo placebo. La calciuria delle 24 ore si è ridotta in media da 390 a 220 mg/die (P < 0.0001) nei trattati con palopegteriparatide, rispetto a una riduzione non significativa nel gruppo placebo (da 329 a 292 mg/die). Alla 52° settimana (20), in cui 78/82 pazienti avevano completato lo studio, l’81% dei pazienti manteneva la calcemia nella norma, mentre il 95% era indipendente dalla terapia convenzionale, e solo 4 pazienti necessitavano ancora di supplementazione di calcio. Il dosaggio iniziale era di 18 µg/die, titolato con incrementi di 3 µg/die fino al raggiungimento della dose ottimale, che si manteneva stabile. Alla 52° settimana la dose media di palopegteriparatide era 24.6 ± 7.6 µg/die. I punteggi SF-36 si sono allineati a quelli della popolazione generale e i questionari specifici per l’ipoparatiroidismo (HPES, Hypoparathyroidism Patient Experience Scale) hanno evidenziato un miglioramento significativo del benessere generale e della qualità della vita in rapporto alla presenza di ipoparatiroidismo.
In un’analisi post-hoc, palopegteriparatide ha mostrato un aumento medio dell’eGFR (+9.3 mL/min/1.73 m² a 52 settimane), con effetto più marcato nei pazienti con eGFR basale < 60 mL/min. Il miglioramento della funzione renale è stato osservato entro 3 mesi dall’inizio del trattamento (21). All’inizio dello studio i pazienti mostravano basso turn-over scheletrico e valori di BMD mediamente elevati. Il trattamento ha indotto riassorbimento scheletrico transitorio, con picco alla 12° settimana, seguito da progressivo riequilibrio. La BMD si è leggermente ridotta nei primi mesi di trattamento per poi stabilizzarsi, avvicinandosi ai valori attesi per età e sesso (20).
Il farmaco è risultato complessivamente ben tollerato; la maggior parte degli eventi avversi è stata di entità lieve o moderata e nessuno ha determinato l’interruzione del trattamento durante la fase in aperto.
Palopegteriparatide ha dimostrato di ripristinare pressochè completamente la funzione fisiologica del PTH nei pazienti con ipoparatiroidismo cronico.
Teriparatide in Italia
Dal 2021 teriparatide PTH(1-34) può essere utilizzato in maniera off-label e senza alcun vincolo temporale in adulti affetti da ipoparatiroidismo cronico, secondo le indicazioni della determinazione (22). In precedenza, era possibile utilizzarlo fino a 36 mesi. Al contrario di palopegteriparatide, nonostante sia un farmaco utilizzato in diversi studi inerenti l’ipoparatiroidismo cronico in bambini e adulti, non ha mai ricevuto un’approvazione ufficiale da EMA o FDA per il trattamento dell’ipoparatiroidismo cronico. È quindi suggerita cautela e comprovata conoscenza ed esperienza negli analoghi del paratormone prima di iniziare questo tipo di trattamento nei pazienti con ipoparatiroidismo cronico.
Posologia iniziale raccomandata: 0.5–0.7 µg/kg/die in 2 somministrazioni sottocutanee, corrispondenti, indicativamente a 20–80 µg/die.
Per il monitoraggio sono previsti prelievi e valutazioni periodiche a cadenze pre-fissate, comprendenti:
- test di gravidanza iniziale;
- esami biochimici: calcemia, fosfatemia, magnesiemia, calciuria 24 h, fosfatasi alcalina ossea, creatinina, transaminasi, elettroliti, emocromo, uricemia, colesterolemia, 25OH-vitamina D, elettroforesi sieroproteica;
- DEXA: individualizzata in base a età e clinica.
Bibliografia
- Orloff LA, Wiseman SM, Bernet VJ, et al. American Thyroid Association statement on postoperative hypoparathyroidism: diagnosis, prevention, and management in adults. Thyroid 2018, 28: 830–41.
- Bilezikian JP. Hypoparathyroidism. J Clin Endocrinol Metab 2020, 105: 1722–36.
- Zavatta G, Clarke BL. Challenges in the management of chronic hypoparathyroidism. Endocr Connect 2020, 9: R229–40.
- Palermo A, Mangiameli G, Tabacco G, et al. PTH(1-34) for the primary prevention of postthyroidectomy hypocalcemia: the THYPOS trial. J Clin Endocrinol Metab 2016, 101: 4039–45.
- Khan AA, Bilezikian JP, Brandi ML, et al. Evaluation and management of hypoparathyroidism: summary statement and guidelines from the second international workshop. J Bone Miner Res 2022, 37: 2568–2585.
- Clarke BL. Hypoparathyroidism: update of guidelines from the 2022 International Task Force. Arch Endocrinol Metab 2022, 66: 604–10.
- Khan S, Khan AA. Hypoparathyroidism: diagnosis, management and emerging therapies. Nat Rev Endocrinol 2025, 31: 360-74.
- Palermo A, Naciu AM, Tabacco G, et al. Calcium citrate: from biochemistry and physiology to clinical applications. Rev Endocr Metab Disord 2019, 20: 353-64.
- Porter RH, Cox BG, Heaney D, et al. Treatment of hypoparathyroid patients with chlorthalidone. N Engl J Med 1978, 298: 577-81.
- Mitchell DM, Regan S, Cooley MR, et al. Long-term follow-up of patients with hypoparathyroidism. J Clin Endocrinol Metab 2012, 97: 4507-14.
- Shao SC, Lai CC, Chen YH, et al. Associations of thiazide use with skin cancers: a systematic review and meta-analysis. BMC Med 2022, 20: 228.
- Kreutz R, Algharably EAH, Douros A. Reviewing the effects of thiazide and thiazide-like diuretics as photosensitizing drugs on the risk of skin cancer. J Hypertens 2019, 37: 1950–8.
- AIFA. Nota informativa importante su idroclorotiazide. 17 ottobre 2018.
- Khan AA, Koch CA, van Uum S, et al. Standards of care for hypoparathyroidism in adults: a Canadian and International Consensus. Eur J Endocrinol 2019, 180: P1–22.
- Palermo A, Naciu AM, Donovan YKT, et al. PTH substitution therapy for chronic hypoparathyroidism: PTH 1–84 and palopegteriparatide. Curr Osteoporos Rep 2025, 23: 12.
- Khan AA, Guyatt G, Ali DS, et al. Management of hypoparathyroidism. J Bone Mineral Res 2022, 37: 2663–77.
- Bollerslev J, et al. European expert consensus on practical management of specific aspects of parathyroid disorders in adults and in pregnancy: recommendations of the ESE educational program of parathyroid disorders. Eur J Endocrinol 2022, 186: R33–63.
- Khan AA, Rejnmark L, Rubin M, et al. PaTH Forward: a randomized, double-blind, placebo-controlled phase 2 trial of TransCon PTH in adult hypoparathyroidism. J Clin Endocrinol Metab 2022, 107: e372-85.
- Khan AA, Rubin MR, Schwarz P, et al. Efficacy and safety of parathyroid hormone replacement with TransCon PTH in hypoparathyroidism: 26-week results from the phase 3 PaTHway trial. J Bone Miner Res 2023, 38: 14-25.
- Clarke BL, Khan AA, Rubin MR, et al. Efficacy and safety of TransCon PTH in adults with hypoparathyroidism: 52-week results from the phase 3 PaTHway Trial. J Clin Endocrinol Metab 2025, 110: 951-60.
- Rejnmark L, Gosmanova EO, Khan AA, et al. Palopegteriparatide treatment improves renal function in adults with chronic hypoparathyroidism: 1-year results from the phase 3 PaTHway trial. Adv Ther 2024, 41: 2500–18.
- Modifica all'allegato 1 della determina n. 1469 del 4 agosto 2017, relativa all'inserimento del medicinale per uso umano teriparatide (Paratormone - PTH) nell'elenco dei medicinali erogabili a totale carico del Servizio sanitario nazionale, ai sensi della legge 23 dicembre 1996, n. 648, quale terapia sostitutiva ormonale per la cura dell'ipoparatiroidismo cronico grave. Determinazione 24 agosto 2021.
- Dettagli
Iperparatiroidismo primario
Inquadramento generale diagnostico delle malattie paratiroidee
Anatomia delle paratiroidi
Ignazio Emmolo
Chirurgia della Tiroide e delle Paratiroidi, Casa di Cura “Città di Bra” – Bra (Cuneo)
Anatomia normale
Le paratiroidi sono quattro, due per lato, distinte in superiori e inferiori. Il 3-15% delle persone presenta ghiandole soprannumerarie, di solito una, ben raramente due, eccezionalmente di più [1,2]. La paratiroide ha consistenza molle e può avere forma diversa: tondeggiante, ovale, allungata, bilobata, a goccia, a fagiolo, a frittella, ecc. Il colore è giallo-bruno più o meno intenso in base alla quantità di cellule adipose presenti dentro la ghiandola [foto A, foto B]. Il grasso può avvolgerla parzialmente o completamente [foto C, foto D]. La dimensione è di 4-5 millimetri, potendo variare in relazione alla forma. Il peso di ogni ghiandola va da 30 a 50 milligrammi, a seconda del sesso e dell’età. Nello stesso soggetto le paratiroidi possono differire tra di loro per dimensione e peso: tutte le ghiandole non pesano più di 200 milligrammi (generalmente intorno a 140 mg)[3].
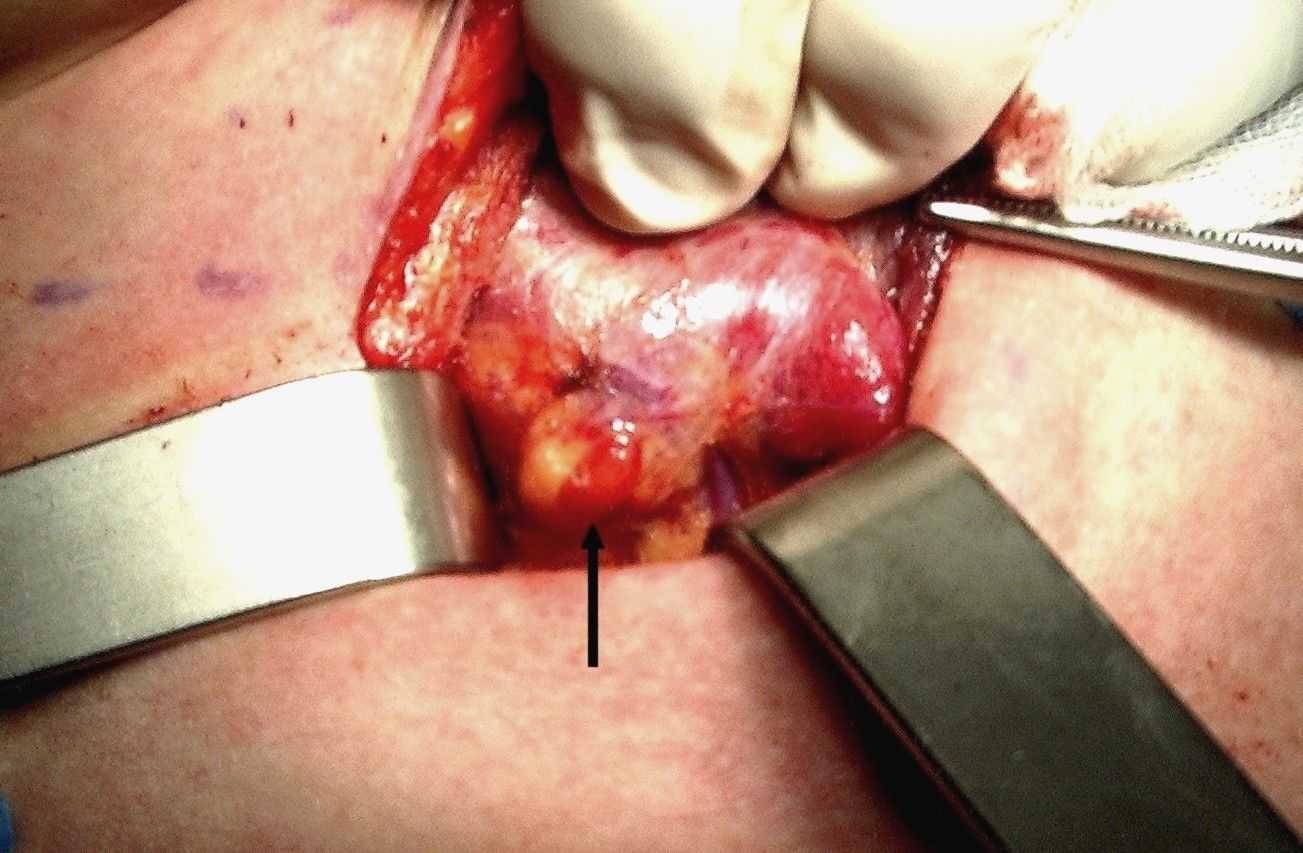
Figura A. Paratiroide normale
La freccia indica la ghiandola situata a ridosso del lobo tiroideo. Essa ha colore bruno intenso che contrasta con il giallo del tessuto adiposo sul quale poggia.
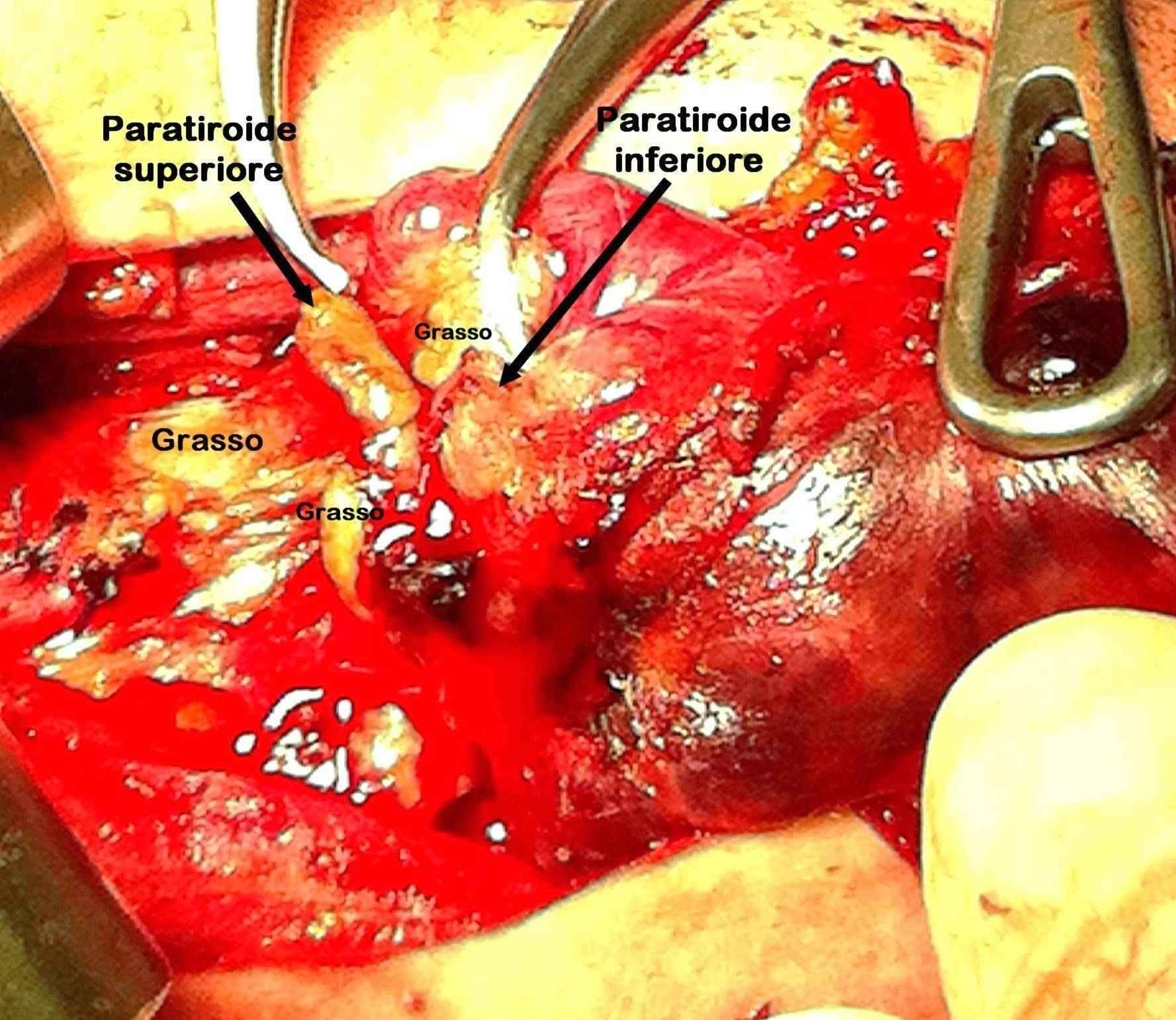
Figura B. Paratiroidi normali
In questa paziente le due paratiroidi hanno colore bruno chiaro e sono poco distinguibili dal tessuto adiposo.
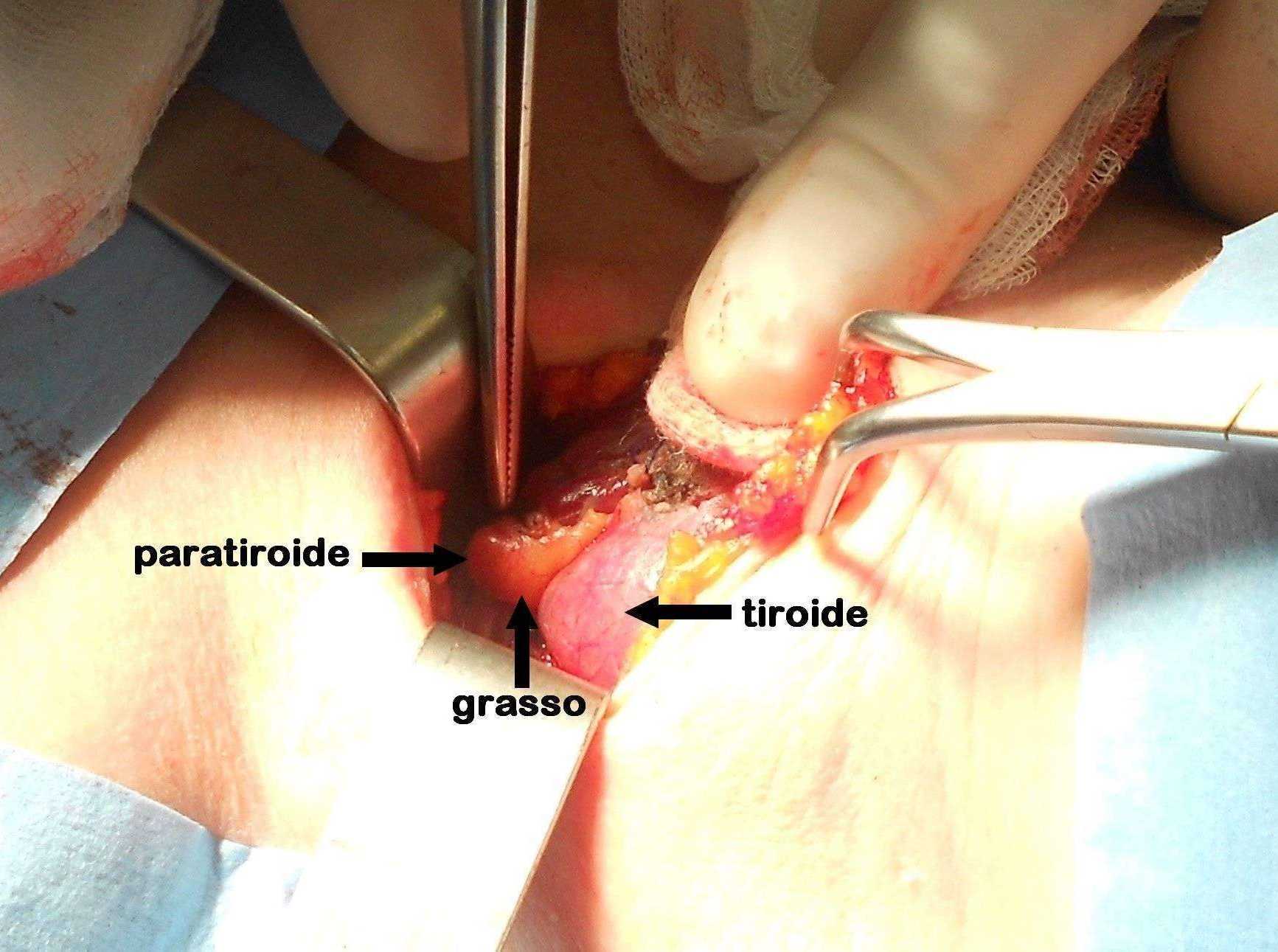
Figura C. Paratiroide parzialmente coperta da grasso
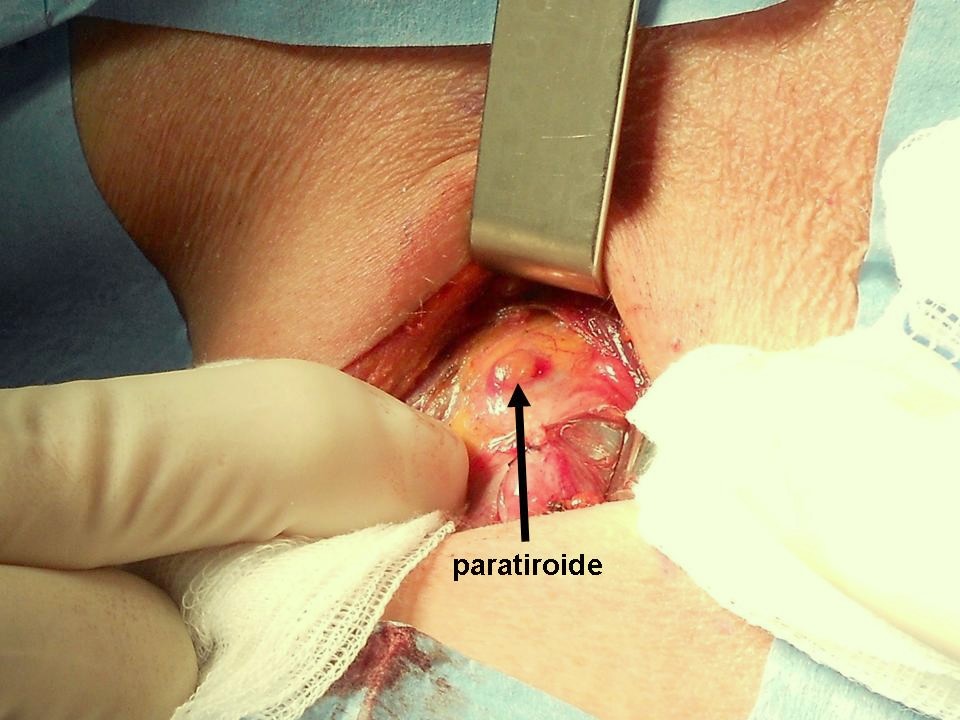
Figura D. Paratiroide completamente coperta da grasso
Un sottile strato di tessuto adiposo ricopre la piccola paratiroide.
L’origine embriologica delle paratiroidi rende conto sia della loro sede di impianto sia delle non rare ectopie [foto E]. Le paratiroidi superiori nascono dalla quarta tasca branchiale assieme alla porzione laterale del lobo tiroideo ed alle cellule parafollicolari. Le inferiori prendono origine dalla terza tasca, assieme al timo. Le inferiori percorrono quindi un tragitto più lungo e, di conseguenza, hanno una posizione meno costante rispetto alle superiori. Queste ultime si trovano di norma sulla superficie posteriore del lobo tiroideo, a livello della porzione medio-superiore [foto F]. Le inferiori hanno invece una sede d’impianto più variabile, il più delle volte vicina al polo tiroideo inferiore o al di sotto di esso [foto G]. In circa l’80% dei casi esiste una simmetria di sede tra i due lati [2]. Nel 15-20% dei soggetti le paratiroidi, specie le inferiori, si trovano in sede ectopica che, in ragione della migrazione embriologica, può essere compresa tra la regione sottomandibolare e quella iuxtapericardica[4]. La sede ectopica delle ghiandole superiori sarà cervicale, mentre quella delle inferiori potrà essere cervicale o mediastinica. Le paratiroidi ectopiche posso trovarsi: sopra il polo tiroideo superiore, dietro il faringe o l’esofago, nella guaina del fascio giugulo-carotideo, all’interno della tiroide, nel cellulare tireo-timico, all’interno del timo [foto H], in sede mediastinica superiore o inferiore, ecc.
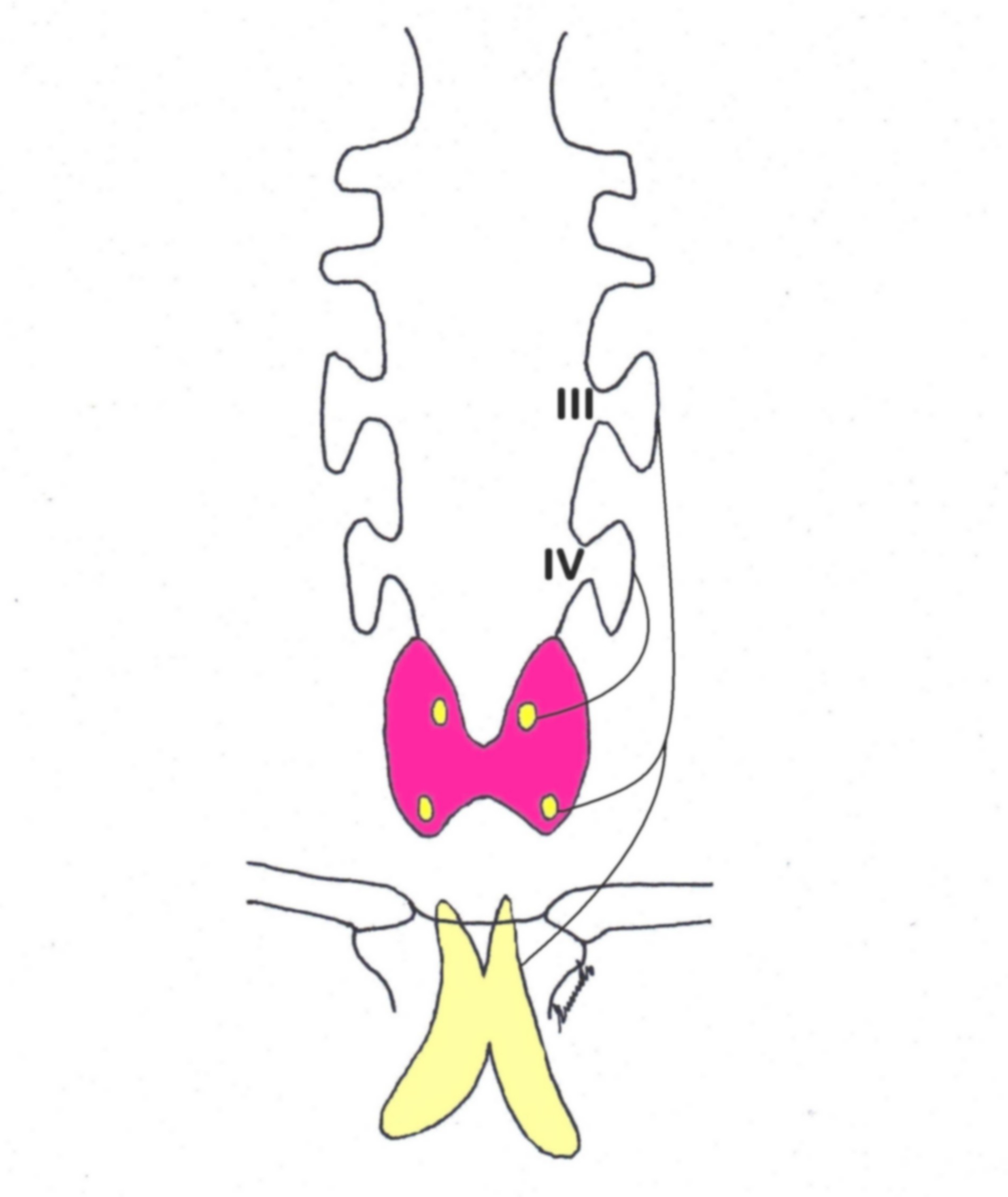
Figura E. Embriologia
Il percorso di migrazione più lungo delle paratiroidi inferiori rende conto della loro sede d'impianto più variabile.
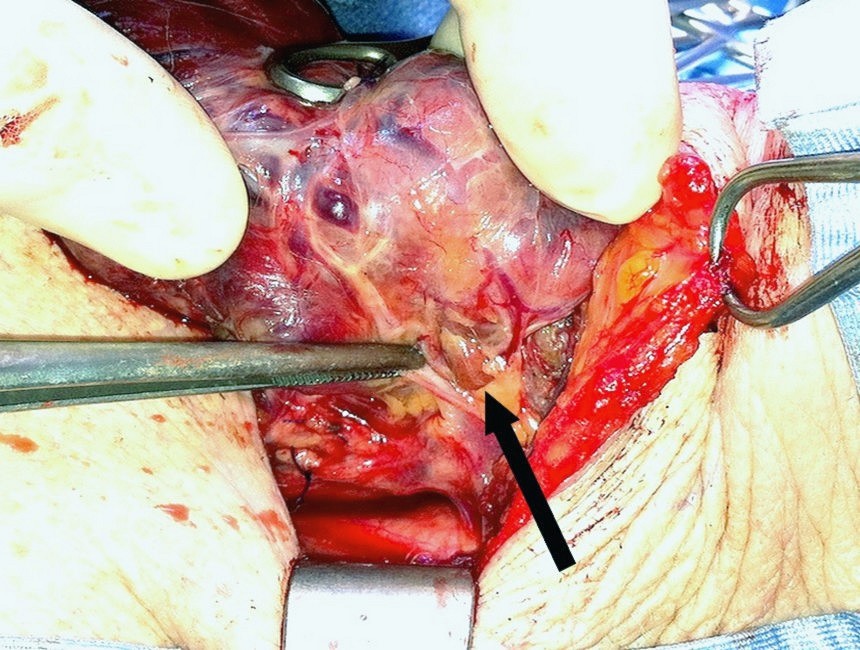
Figura F. Sede della paratiroide superiore
La paratiroide superiore sinistra normale nella sua sede tipica
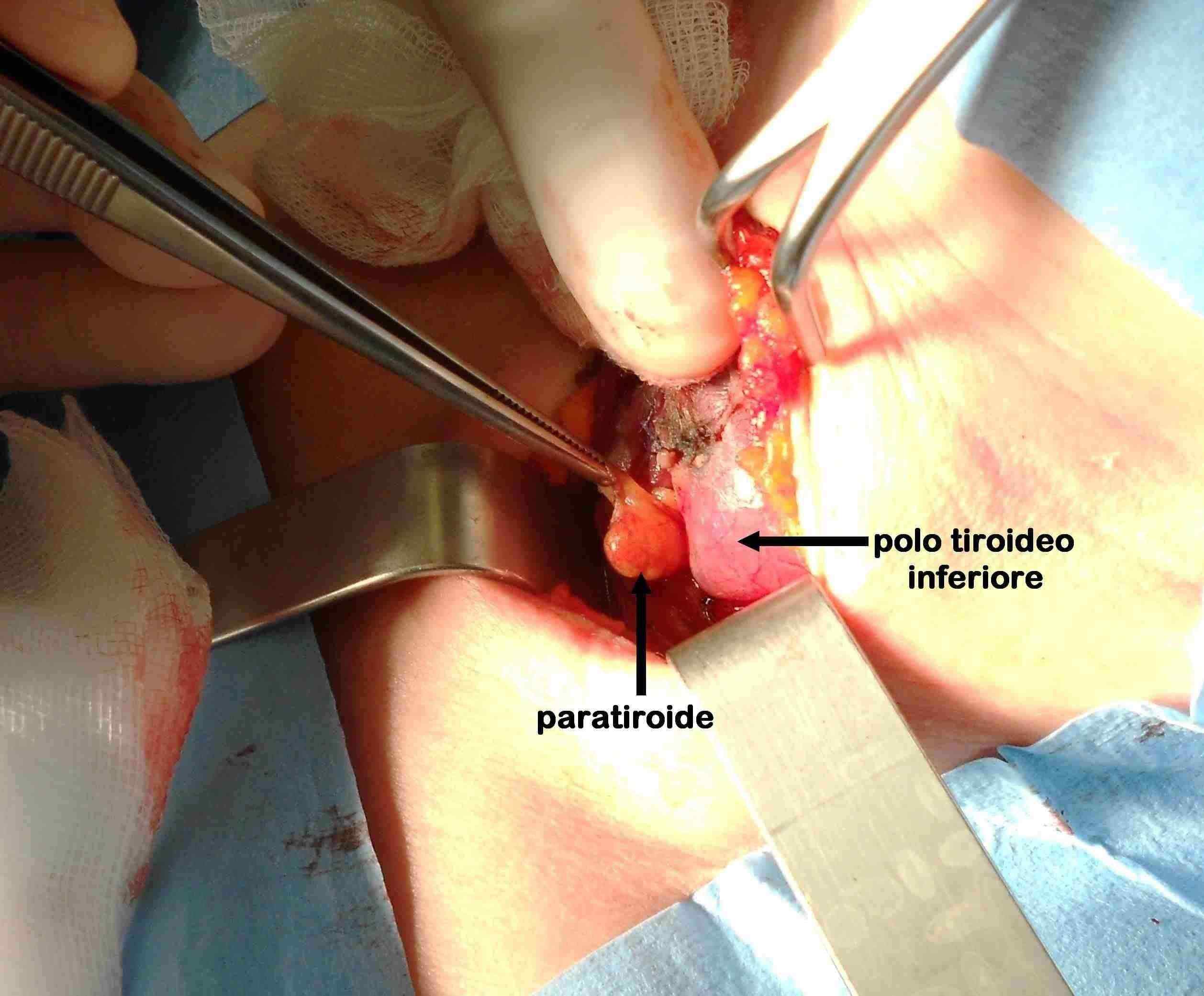
Figura G. Sede della paratiroide inferiore
La paratiroide inferiore sinistra normale subito al di sotto del polo tiroideo inferiore.
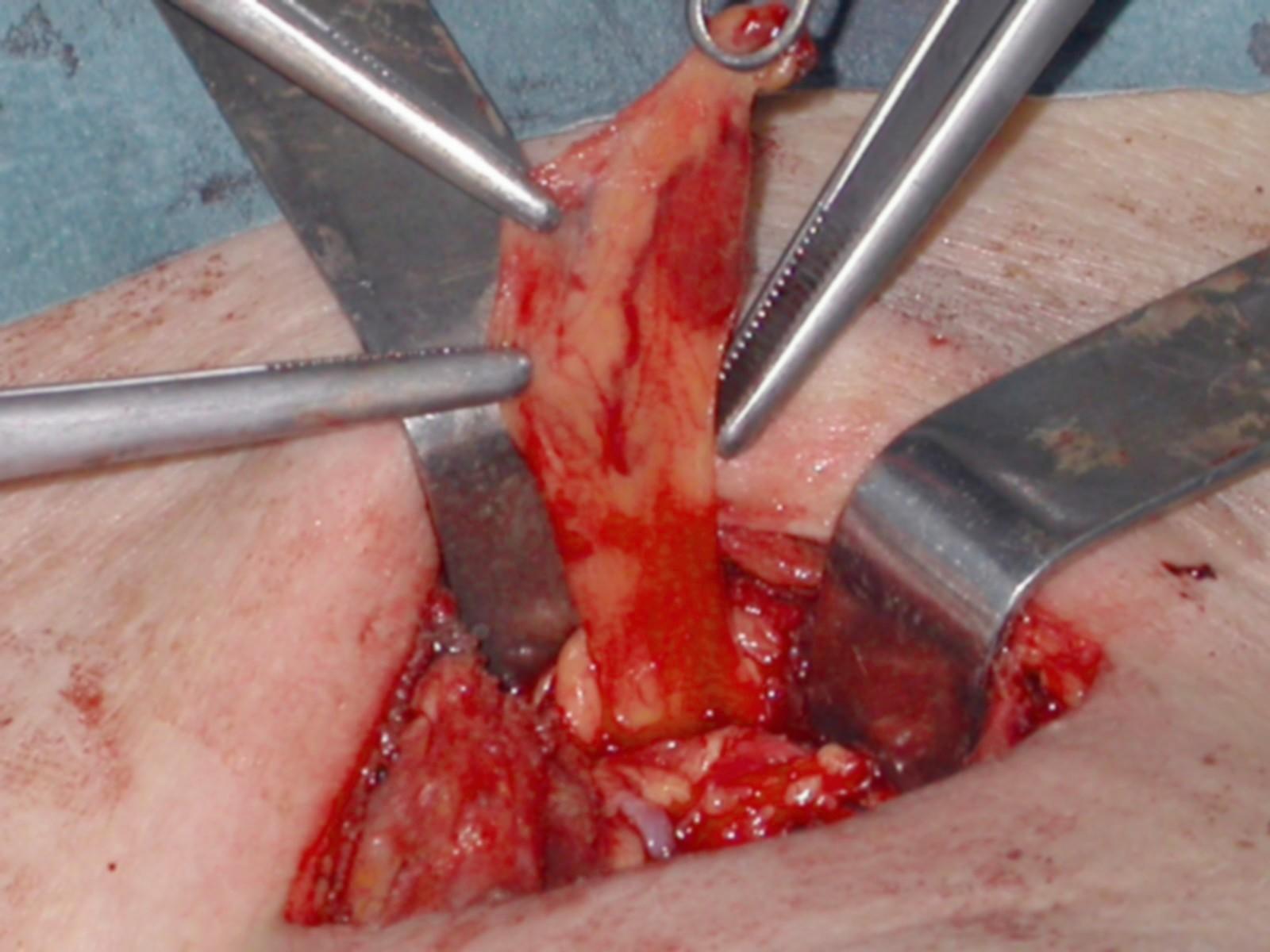
Figura H. Piccolo adenoma paratiroideo ectopico intra-timico
Il corno timico superiore è stato mobilizzato ed attratto fuori dal mediastino. Al suo interno traspare, per il colore più scuro, un piccolo adenoma accompagnato dal vaso nutritizio.
Le paratiroidi sono costituite da due tipi di cellule parenchimali[5][foto I]:
- cellule principali, di piccole dimensioni, sono in numero predominante e secernono il PTH,
- cellule ossifile, di dimensioni maggiori, sono ricche di mitocondri e hanno significato biologico incerto.
Le cellule chiare, considerate una variante delle principali, sono presenti normalmente nell’infanzia.
All’interno delle paratiroidi sono contenute anche cellule adipose in quantità inversamente proporzionale all’attività secretoria ghiandolare.
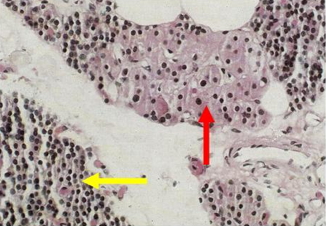
Figura I. Istologia della paratiroide normale
La freccia gialla indica le cellule principali e la freccia rossa quelle ossifile. Queste ultime sono più grandi per il loro abbondante citoplasma ricco di mitocondri.
Anatomia patologica
L’iperparatiroidismo primitivo può essere dovuto a
- adenoma (circa 85% dei casi, fra i quali multiplo nel 2-5%),
- iperplasia (circa 15% dei casi),
- carcinoma (< 1% dei casi).
Negli ultimi anni alcuni autori hanno riportato una maggiore incidenza dell’adenoma singolo (90%) e minore dell’iperplasia (6%) [6].
L’iperparatiroidismo secondario è invece sostenuto sempre dall’iperplasia.
L’adenoma è in genere di forma nodulare, capsulato, di dimensioni variabili [foto L, foto M]]. Il colore è più scuro rispetto alla ghiandola normale, di solito compreso tra il bruno-rossastro ed il rosso-vinoso. Esso è riccamente costituito da cellule principali, più raramente ossifile o chiare, con scarsa o minima presenza di cellule adipose. Talvolta in periferia è presente un orletto più chiaro costituito da tessuto paratiroideo normale compresso dalla neoplasia. Esso risulta determinante per porre la diagnosi differenziale con l’iperplasia, specialmente all’esame istologico estemporaneo. Quest’ultimo può incontrare notevole difficoltà nel distinguere tra natura paratiroidea e tiroidea di un nodo con estesa proliferazione di cellule ossifile.
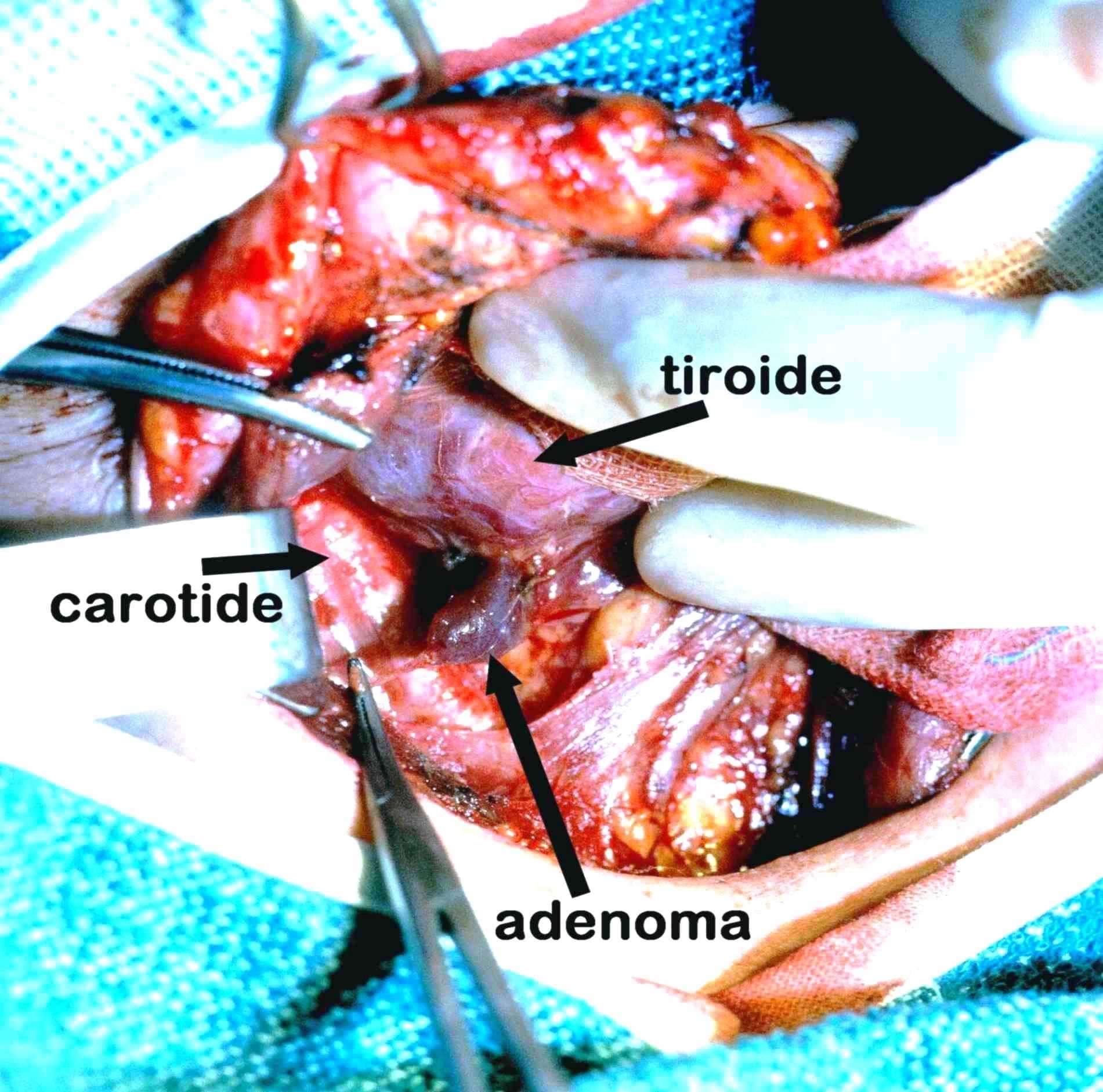
Foto L. Piccolo adenoma paratiroideo
L'adenoma di 12 mm, di colore rosso scuro, è adeso al lobo tiroideo.
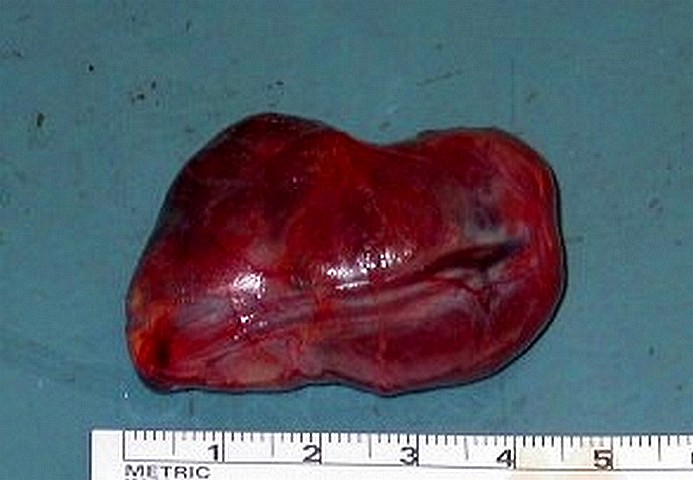
Foto M. Grosso adenoma paratiroideo
Nonostante le cospicue dimensioni la neoplasia ha caratteristiche macroscopiche di benignità, confermata dall'esame istologico.
L’iperplasia interessa tutte le ghiandole che, per il differente grado di coinvolgimento, raramente hanno la stessa dimensione, potendo una apparire di volume anche molto superiore alle altre. La proliferazione cellulare può essere di tipo diffuso o nodulare. Non è possibile distinguere istologicamente tra iperplasia primitiva e secondaria. La diagnosi differenziale intra-operatoria (macroscopica ed istologica estemporanea) tra iperplasia ed adenoma è in genere poco affidabile con l’esame di una sola ghiandola, a meno del riscontro dell’orletto di tessuto normale (vedi sopra). Tuttavia l’esame di un’altra paratiroide permette di differenziare la malattia multi-ghiandolare da quella uni-ghiandolare.
Il carcinoma ha il più delle volte grosse dimensioni, consistenza dura, ed è aderente alle strutture contigue. Le caratteristiche macroscopiche, la presenza di atipie cellulari, l’elevata attività mitotica non costituiscono criteri di certezza per la diagnosi differenziale tra carcinoma e adenoma. Sono invece criteri di certezza l’invasione dei vasi, della capsula, dei tessuti circostanti e la presenza di metastasi a distanza, in assenza dei quali si preferisce porre diagnosi di adenoma atipico[7].
Bibliografia
- Wang C. The anatomic basis of parathyroid surgery. Ann Surg 1976, 183: 271-5.
- Akerström G, Malmaeus J, Bergström R. Surgical anatomy of human parathyroid glands. Surgery 1984, 95: 14-21.
- Testut L, Jacob O. Trattato di anatomia topografica. Unione Tipografico-Editrice Torinese 1974, vol II: 84-7.
- Yeo H, Uranga P, Roman S. Conventional surgical management of primary hyperparathyroidism. In Oertli D, Udelsman R. Surgery of the thyroid and parathyroid glands. Berlin: Springer-Verlag, 2007: 261-7.
- Lewis PD. Surgical Pathology of the parathyroids in primary and secondary hyperparathyroidism. In Lynn J, Bloom SR. Surgical Endocrinology. Butterworth-Heinemann Ltd, Oxford, 1993: 370-9.
- Ruda Jm, Hollenbeak CS, Stack BC. A systematic review of the diagnosis and treatment of primary hyperparathyroidism from 1995 to 2003. Otolaryngol Head Neck Surg 2005, 132: 359-72.
- Adam MA, Untch BR, Olson JA. Parathyroid carcinoma: current understanding and new insights into gene expression and intraoperative parathyroid hormone kinetics. The Oncologist 2010, 15: 61-72.