Disfunzione erettile
Terapia farmacologica del craniofaringioma

Marco Faustini Fustini
Scuola AME Malattie Ipotalamo-Ipofisarie, Bologna
(aggiornamento 4/5/2026)
Introduzione
L’impiego di farmaci nella terapia del craniofaringioma è stato a lungo limitato all’impiego di iniezioni intra-cistiche di bleomicina o alfa-interferone nei pazienti pediatrici, volte principalmente a ritardare la progressione di malattia e permettere un successivo trattamento chirurgico e radiante (1,2).
L’identificazione di mutazioni somatiche, potenzialmente in grado di aprire nuovi scenari nella terapia farmacologica sistemica del craniofaringioma, è storia relativamente recente. Fin dall’inizio degli anni 2000, nel 65-70% di pazienti con la variante adamantinomatosa – che comprende complessivamente il 60-80% dei casi di craniofaringioma - furono identificate mutazioni nell’esone 3 del gene CTNNB1, inattivanti l’attività enzimatica GSK-3b (glycogen synthase kinase-3b) all’interno della via WNT, con conseguente ridotta degradazione e accumulo di ß-catenina e sua traslocazione nel nucleo (3,4). Come è noto, ß-catenina induce la trascrizione di geni e fattori di crescita che stimolano la proliferazione cellulare. Al contrario, fino a non molti anni fa non erano state ancora identificate mutazioni responsabili dello sviluppo della variante papillare.
Il punto di svolta nella possibile terapia farmacologica del craniofaringioma ci fu nel 2014, con il sequenziamento dell’intero esoma di 12 craniofaringiomi adamantinomatosi e di 3 craniofaringiomi papillari (di cui uno in un bambino di 9 anni), mediante una nuova tecnica altamente specifica che permetteva di identificare mutazioni somatiche anche con una frazione allelica molto bassa - assai comune in popolazioni cellulari eterogenee come si reperta spesso nei craniofaringiomi, dove la contaminazione con cellule stromali e tessuto reattivo è la regola (5). A conferma di studi precedenti, il gene più frequentemente mutato nei craniofaringiomi adamantinomatosi fu CTNNB1, esclusivamente nell’esone 3, in 11/12 casi (92%). Tutti e tre i craniofaringiomi papillari (100%) mostrarono mutazioni (c.1799T>A) dell’oncogene BRAFV600E, attivanti in maniera costitutiva la serin-treonin-chinasi, che regola la via MAPK/ERK coinvolta nella divisione e nella differenziazione cellulare. Questi dati furono validati nello stesso studio mediante analisi di genotipizzazione indirizzate sui bersagli individuati dal sequenziamento dell’esoma su 98 campioni di tessuti estratti da 95 pazienti (36 con variante papillare e 59 con variante adamantinomatosa): mutazioni di CTNNB1 furono individuate nel 96% dei campioni di craniofaringioma adamantinomatoso, ma in nessuno di questi fu individuata la mutazione BRAFV600E, che, viceversa, fu evidenziata nel 95% dei craniofaringiomi papillari, senza che questi mostrassero mutazioni di CTNNB1. La conclusione fu che i craniofaringiomi adamantinomatosi e i craniofaringiomi papillari mostravano mutazioni clonali mutuamente esclusive (5).
Terapia farmacologica del craniofaringioma papillare
La scoperta della mutazione BRAFV600E nei craniofaringiomi papillari suscitò subito un certo interesse anche in campo diagnostico, in particolare sui campioni tissutali scarsi ottenuti da lesioni cistiche, in cui la distinzione con la cisti della tasca di Rathke non è sempre agevole con le comuni metodiche di immuno-istochimica (6,7). Non vi è dubbio, tuttavia, che l’impatto clinico prevalente si ebbe successivamente, allorchè si aprirono nuove prospettive nella terapia farmacologica dei craniofaringiomi papillari. Mutuando l’esperienza maturata in altre neoplasie con mutazioni BRAFV600E (8-14), nel 2016 fu pubblicato il risultato della terapia con la doppia inibizione di BRAF (dabrafenib) e MEK (trametinib) in un paziente con craniofaringioma papillare BRAFV600E-mutato, in precedenza sottoposto a numerosi interventi neurochirurgici (15). Il paziente mostrò una risposta eccellente in termini di riduzione volumetrica sia della componente solida (85%) sia di quella cistica (81%). Altri autori proposero l’impiego del solo inibitore di BRAF verumafenib, che però risultava molto efficace durante il trattamento, ma gravato da rapida ricrescita a breve termine dopo la sospensione del farmaco rispetto al trattamento con la doppia inibizione BRAF/MEK (16). La possibile spiegazione è che in altre neoplasie BRAF-mutate, in cui la maggior parte dei fenomeni di resistenza al singolo inibitore di BRAF avviene per la riattivazione della via MAPK (RAF-MEK-ERK), l’aggiunta di un inibitore MEK ritarda la comparsa di cloni resistenti, oltre a ridurre lo sviluppo di carcinomi cutanei squamo-cellulari, complicanza più frequente con l’impiego del solo inibitore di BRAF (9).
Dopo le iniziali segnalazioni come singoli case report e piccole serie (15-17), furono disegnati studi in popolazioni più vaste di pazienti con craniofaringioma papillare BRAF-mutato. Uno studio prospettico (18) valutò i risultati della terapia combinata con inibitore di BRAF (vemurafenib) e inibitore di MEK (cobimetinib) a distanza di almeno tre settimane dall’intervento chirurgico in 16 adulti di nuova diagnosi, che non erano stati preventivamente trattati con radioterapia o terapie sistemiche e che avevano un residuo di neoplasia di almeno 10 mm di diametro (volume mediano 2.75 cm3). Ogni ciclo terapeutico di 28 giorni prevedeva la somministrazione di vemurafenib per tutta la durata del ciclo e di cobimetinib per 21 giorni. Dopo una mediana di 8 cicli, 15/16 pazienti (94%) mostrarono una significativa riduzione volumetrica della neoplasia (mediana 91%). L’unico paziente che non aveva raggiunto una risposta volumetrica, in realtà non era riuscito a completare neppure il primo ciclo, per la comparsa di seri effetti collaterali. La mediana del follow-up fu di 22 mesi e la percentuale stimata di pazienti che continuavano ad avere una risposta volumetrica a 12 mesi era del 93%. Tre dei 15 pazienti che avevano ottenuto una buona riduzione volumetrica avevano sviluppato progressione di malattia dopo che la terapia era stata interrotta. Complessivamente, 8 pazienti ricevettero altri trattamenti (radioterapia, secondo approccio chirurgico, inibitore di BRAF: dabrafenib) dopo l’interruzione dei cicli di terapia combinata con inibitori BRAF-MEK e 7 pazienti non ricevettero alcun trattamento: dopo follow-up mediano di 23 mesi, sei non mostrarono progressione. Ci furono 12 eventi avversi di grado 3 (eruzione cutanea, ipertensione arteriosa, disidratazione, prolungamento QT, reazioni allergiche, aumento di fosfatasi alcalina, iponatriemia) e due eventi avversi di grado 4 (aumento di CPK e iperglicemia), con necessità di interruzione del trattamento in tre casi.
Un successivo studio multicentrico di coorte reclutò retrospettivamente 16 pazienti con craniofaringioma papillare BRAF-mutato, trattati tra il 2019 e il 2023 con la doppia inibizione BRAF-MEK con l’associazione dabrafenib-trametinib (19). La popolazione era eterogenea: terapia neo-adiuvante (in 6 pazienti di nuova diagnosi con tumori voluminosi, in cui non era proponibile l’intervento neurochirurgico), terapia adiuvante (in 8 pazienti già sottoposti a procedure chirurgiche singole o multiple prima della terapia radiante) o terapia palliativa a lungo termine (in due pazienti che avevano sviluppato recidive o con residui tumorali rilevanti nonostante precedenti trattamenti multi-modali). Anche in questo studio il 94% dei pazienti (15/16) mostrò evidente riduzione volumetrica (subtotale o parziale): in media 81.4 ± 18.3% all’ultimo controllo. La terapia fu ben tollerata in 10 pazienti (62.5%); gli eventi avversi (pneumopatia ipossiemica, febbre, incremento degli indici di citolisi epatica, cefalea, vomito, astenia, eruzione cutanea, edemi periferici, diarrea, mialgie) portarono all’interruzione temporanea del trattamento in 5 pazienti (grado 1 e 3) e definitiva in tre pazienti (grado 3).
Se da un lato il trattamento mediante la doppia inibizione BRAF-MEK nei craniofaringiomi papillari ha indubbiamente aperto nuove prospettive nella gestione di questi tumori, dall’altro rimangono ancora da chiarire alcuni punti cruciali: la durata del trattamento farmacologico, il momento ideale in cui eventualmente proporlo al paziente durante il suo percorso terapeutico, l’eventuale modificazione del work-up diagnostico-terapeutico. Al momento, non siamo in grado di formulare una diagnosi pre-operatoria di craniofaringioma papillare solamente sulla base della storia clinica, dell’età del paziente e dell’imaging. Pertanto, il team multi-disciplinare (comprendente il patologo, l’endocrinologo, il neurochirurgo, il neuroradiologo, il radioterapista e l’oncologo) rimane fondamentale nel definire il work-up diagnostico e la migliore strategia terapeutica.
Terapia farmacologica del craniofaringioma adamantinomatoso
Il coinvolgimento della via WNT/ß-catenina nella tumorigenesi della variante adamantinomatosa potrebbe rappresentare un primo passo verso l’identificazione di una terapia mirata su un preciso bersaglio molecolare (3,4). Tuttavia, la complessità della via e l’incompleta conoscenza dei meccanismi fisiologici che la regolano rendono attualmente ancora prematura l’identificazione di una terapia mirata su questo bersaglio. Comunque, è meritevole di attenzione una recente scoperta che potrebbe fornire ulteriori elementi di speculazione in termini di prospettive terapeutiche, in particolare per i casi gravati da frequenti recidive e ripresa di malattia (20,21). Va aggiunto, peraltro, che le rare forme di craniofaringioma maligno descritte in letteratura derivano dalla variante adamantinomatosa in oltre i due terzi dei casi (22).
A differenza della variante papillare, in cui la via MAPK/ERK è attivata come conseguenza della mutazione somatica dell’oncogene BRAFV600E, quella adamantinomatosa non mostra generalmente l’attivazione di questa via. Tuttavia, alcuni dati mostrano che l’attivazione della via MAPK/ERK può avvenire in maniera paracrina in alcune aree di craniofaringioma adamantinomatoso, soprattutto nelle forme gravate da maggiore aggressività, in seguito all’espressione locale di fattori multipli, quali alcuni membri della famiglia FGF e EGF, come pure di ligandi e proteine trans-membrana (ad esempio CD47) (20,23). Sulla scorta di questi dati, un recente studio pre-clinico ha mostrato che in 6/14 casi di craniofaringioma adamantinomatoso recidivante era evidente l’attivazione della via MAPK/ERK nelle aree prospicienti il fronte di aggressività locale, fornendo così il presupposto per futuri studi clinici per valutare l’eventuale impiego di inibitori della via MAPK nei craniofaringiomi adamantinomatosi aggressivi e recidivanti (21).
Bibliografia
- Zheng S, Fang Y, Cai BW, et al. Intracystic bleomycin for cystic craniopharyngiomas in children. Cochrane Database Syst Rev 2016, 7: CD008890.
- Kilday J-P, Caldarelli M, Massimi L, et al. Intracystic interferon-alpha in pediatric craniopharyngioma patients: an international multicenter assessment on behalf of SIOPE and ISPN. Neuro-Oncology 2017, 19: 1398-407.
- Hofmann BM, Kreutzer J, Saeger W, et al. Nuclear ß-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and Rathke cleft cysts: a clinico-pathologic approach. Am J Surg Pathol 2006, 30: 1595-603.
- Oikonomou E, Barreto DC, Soares B, et al. ß-catenin mutations in craniopharyngiomas and pituitary adenomas. J Neurooncol 2005, 73: 205-9.
- Brastianos PK, Taylor-Weiner A, Manley PE, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet 2014, 46: 161-5.
- Marucci G, de Biase D, Zoli M, et al. Targeted BRAF and CTNNB1 next-generation sequencing allows proper classification of nonadenomatous lesions of the sellar region in samples with limiting amounts of lesional cells. Pituitary 2015, 18: 905-11.
- Kim JH, , Pailus W, Heim S. BRAF V600E mutation is a useful marker for differentiating Rathke’s cleft cyst with squamous metaplasia from papillary craniopharyngioma. J Neurooncol 2015, 123: 189-91.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010, 363: 809-19.
- Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhinition in melanoma with BRAF V600 mutations. N Engl J Med 2012, 367: 1694-703.
- Hyman DM, PuzanovI, Subbiah V, et al. Vemurafenib in multiple nonmelanoma. cancers with BRAF V600 mutations. N Engl J Med 2015, 373: 726-36.
- Dietrich S, Glimm H, Andrulis M, et al. BRAF inhibition in refractory hairy-cell leukemia. N Engl J Med 2012, 366: 2038-40.
- Munoiz J, Schlette E, Kurzrock R. Rapid response to vemurafenib in a heavely pretreated patient with hairy cell leukemia and BRAF mutation. J Clin Oncol 2013, 31: e351-2.
- Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAFV600E-mutated Erdheim-Chester disease. J Clin Oncol 2014, 33: 411-8.
- Kaye FJ, Ivey AM, Drane WE, et al. Clinical and radiographic response with combined BRAF-targeted therapy in stage 4 ameloblastoma. J Natl Cancer Inst 2015, 107: 378.
- Brastianos PK, Shankar GM, Gill CM, et al. Dramatic response of BRAF V600E mutant papillary craniopharyngioma to targeted therapy. J Natl Cancer Inst 2016, 188: djv310.
- Aylwin SJ, Bodi I, Beaney R. Pronounced responce of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary 2016, 19: 544-6.
- Bernstein A, Mrowczynski OD, Greene A, et al. Dual BRAF/MEK therapy in BRAF V600E-mutated primary brain tumors: a case series showing dramatic clinical and radiographic responses and a reduction in cutaneous toxicity. J Neurosurg 2019, 133: 1704-9.
- Brastianos PK, Twohy E, Geyer S, et al. BRAF-MEK inhibition in newly diagnosed papillary craniopharyngiomas. N Engl J Med 2023, 389: 118-26.
- De Alcubierre D, Gkasdaris G, Mordrel M, et al. BRAF and MEK inhibitor targeted therapy in papillary craniopharyngiomas: a cohort study. Eur J Endocrinol 2024, 191: 251-61.
- Apps JR, Carreno G, Gonzales-Meljem JM, et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol 2018, 135: 757-77.
- Apps JR, Gonzales-Meljem JM, Guiho R, et al. Recurrent adamantinomatous craniopharyngiomas show MAPK pathway activation, clonal evolution and rare TP53-loss-mediated malignant progression. Acta Neuropathol Commun 2024, 12: 127.
- Wang F, He Y, Wang Y, et al. Malignant craniopharingioma: a report of seven cases and review of the literature. World Neurosurg 2020, 135: e194-201.
- Zhang H, Wang C, Fan J, et al. CD47 promotes the proliferation and migration of adamantinomatous craniopharyngioma cells by activating the MAPK/ERK pathway, and CD47 blockade facilitates microglia-mediated phagocytosis. Neuropathol Appl Neurobiol 2022, 48: e12795.
Inquadramento generale diagnostico delle malattie ipotalamo-ipofisarie
Fisiologia ipotalamo-ipofisaria
Asse TRH-TSH-tiroide
Asse CRH-ACTH-surrene
Asse GHRH-GH-IGF
Asse GnRH-gonadotropine-gonade femminile
Anatomia chirurgica della regione sellare e sovrasellare
Giovanni Lasio, Martina Revay
Neurochirurgia, Istituto Clinico Humanitas, Rozzano (MI)
L'osso sfenoide, situato al centro della base cranica, è in stretto rapporto con le cavità nasali inferiormente e con l'ipofisi superiormente, contenuta nella struttura ossea, detta sella, situata centralmente nel corpo dello sfenoide e che aggetta nel seno sfenoidale. I nervi olfattori, il giro retto, la parte posteriore del lobo frontale, il pavimento del III ventricolo, il ponte, il mesencefalo ed il chiasma ottico sono le strutture nervose a più stretto contatto con lo sfenoide. I nervi cranici dal II al VI sono pure in stretti rapporti con l'osso sfenoide, in quanto i forami da cui escono/entrano nel cranio sono tutti localizzati nell'osso sfenoide stesso: canale ottico (II), fessura orbitaria superiore (III, IV, VI e branca orbitaria del V), forame rotondo (branca mascellare del V), forame ovale (branca mandibolare del V).
Oltre che importanti rapporti con strutture nervose, l'osso sfenoide ha intimi rapporti con strutture vascolari arteriose e venose. Le arterie carotidi attraversano lo sfenoide prima di entrare nel cranio, l'arteria basilare è addossata alla sua faccia posteriore, mentre il poligono di Willis è situato al di sopra della sua porzione centrale. I seni cavernosi sono appoggiati all'osso sfenoide, che ne forma la parete laterale, mentre la dura sellare ne forma la parete mediale; protrudono all'interno del seno sfenoidale. I seni cavernosi sono uniti dai seni inter-cavernoso superiore ed inferiore, che delineano la parete anteriore della sella e la parete inferiore. Il seno basilare poi connette i seni cavernosi posteriormente al dorsum sellae ed è usualmente il seno di maggiori dimensioni. I seni petrosi inferiori e superiori si uniscono al seno basilare.
La parte successiva è divisa in 3 sezioni: anatomia della regione sellare propriamente detta (vista dal basso) e della regione soprasellare (vista dall'alto), anatomia della ghiandola ipofisaria.
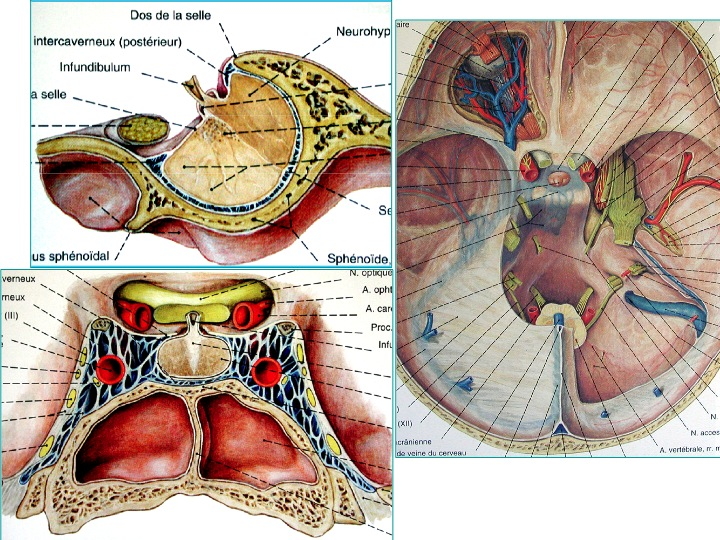
Anatomia della regione sellare in proiezione sagittale (in alto a sinistra), coronale dal basso (in basso a sinistra) e assiale dall' alto (a destra).
Anatomia della regione sellare propriamente detta (vista dal basso)
Il corpo dello sfenoide contiene il seno sfenoidale, che è in comunicazione con le cavità nasali tramite due osti, simmetrici. Il seno sfenoidale è soggetto a una considerevole variabilità di dimensione, forma e pneumatizzazione. Nell'adulto esistono tre tipi di seno sfenoidale: concale, presellare e sellare, così definiti in base alla pneumatizzazione, assente nel tipo concale e ben presente nel tipo sellare (75%). Il seno sfenoidale è poi attraversato da setti, assai variabili come numero, forma, direzione, spessore, ecc. I canali ottici protrudono nella porzione supero-laterale del seno sfenoidale, mentre la II branca del trigemino protrude nella sua parte infero-laterale. Il recesso ottico-carotideo è situato lateralmente e superiormente fra canale ottico e protuberanza carotidea.
La struttura ossea definita sella turcica, contenuta nella parte centrale del corpo dell' osso sfenoide, sporge all'interno del seno sfenoidale e ha confini anatomici precisi:
- postero-superiormente il dorso della sella, il cui margine libero termina lateralmente con i processi clinoidei posteriori e si continua inferiormente nel clivus;
- i seni cavernosi, che contengono sangue venoso, sono situati lateralmente alla sella. All'interno di essi decorre la porzione orizzontale della carotide interna ed un segmento del VI nervo cranico, mentre il III, il IV e la I branca del V nervo cranico si trovano nel tetto e nella parete laterale del seno cavernoso. Dal segmento intra-cavernoso della carotide nascono l'arteria meningo-ipofisaria, l'arteria del seno cavernoso inferiore e le arterie capsulari. La parete mediale del seno cavernoso è costituita da dura sottile, ma può presentare dei buchi od essere del tutto assente. La distanza fra arteria carotide e faccia laterale dell'ipofisi in condizioni normali varia fra 1 e 3 mm;
- anteriormente e superiormente il tubercolo della sella e il piano sfeno-etmoidale, separati dal solco chiasmatico;
- inferiormente il pavimento osseo della sella, che si continua con il clivus. Anteriormente la parete anteriore della sella, il clivus e la parete anteriore ossea dei seni cavernosi aggettano nella cavità del seno sfenoidale;
- il tetto della sella è costituito dal diaframma sellare, un setto orizzontale di dura che divide la loggia sellare dalla regione sovra-sellare, con un foro centrale per il passaggio del peduncolo ipofisario, struttura che connette l’ipotalamo all’ipofisi. Il diaframma è più sottile attorno al peduncolo e la sua apertura centrale è di solito più ampia di quanto necessario per il passaggio del peduncolo. In questi casi è presente un'invaginazione dell' aracnoide sovrasellare. La sella è internamente rivestita da periostio e contiene l'ipofisi.
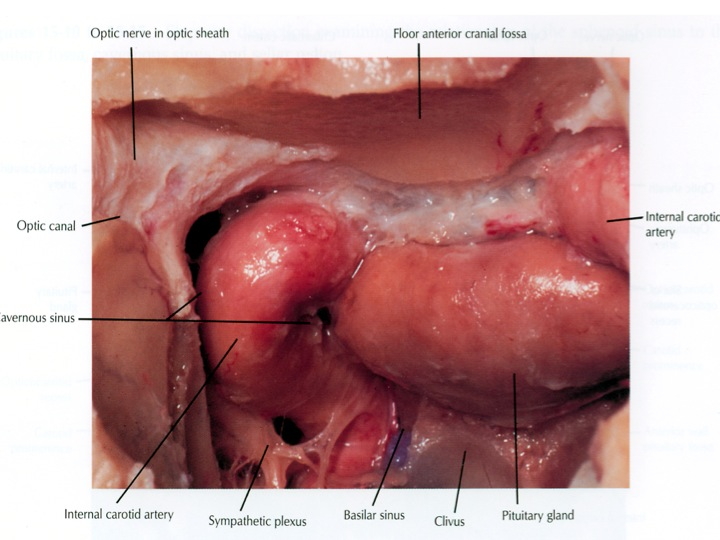
Visione endoscopica della sella, seno sfenoidale e carotidi clivali e cavernose dall’accesso TNS
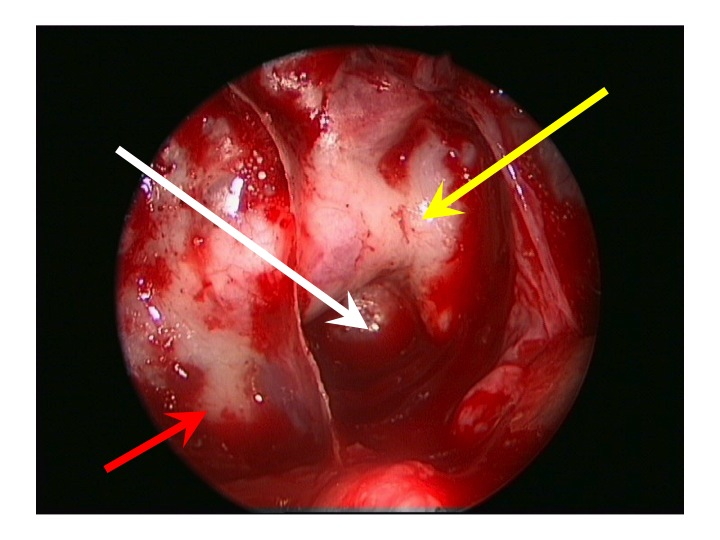
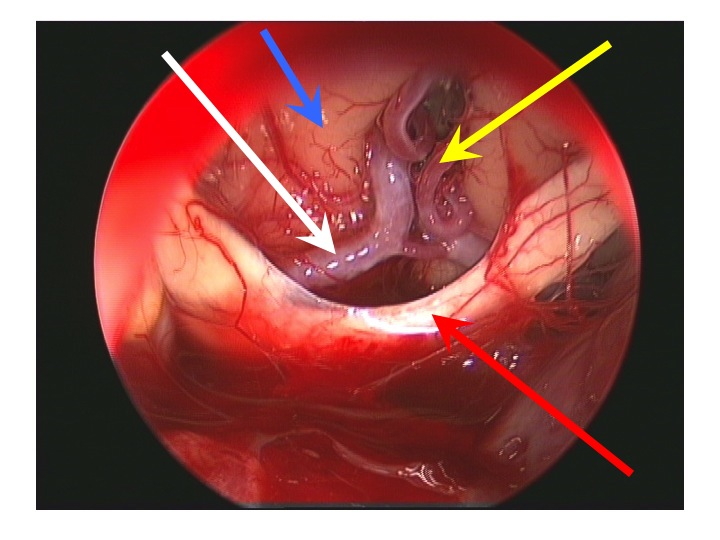
Visione endoscopica della regione sovrasellare (via TNS extended).
Nell'immagine in alto, la freccia rossa indica la carotide clivale dx, la gialla il seno cavernoso sinistro, la bianca attraversa la sella e indica il clivus.
Nell'immagine in basso, la freccia rossa indica il bordo anteriore del chiasma, la gialla l'arteria di Heubner sinistra, la bianca il tratto A1 della cerebrale anteriore dx, la blu la faccia inferiore del lobo frontale dx.
Anatomia della regione soprasellare (vista dall'alto)
La regione sovrasellare ha limiti più imprecisi. Può essere definita come la regione delimitata:
- anteriormente dal tubercolo sellare;
- inferiormente dal diaframma;
- posteriormente dalla membrana di Lillequist, una membrana aracnoidea che decorre fra il dorso della sella e la faccia anteriore dei corpi mammillari e che separa la cisterna chiasmatica dalla cisterna inter-peduncolare;
- superiormente dai nervi ottici, dal chiasma e dal pavimento del III ventricolo;
- lateralmente dalle carotidi.
Nervi e vasi della regione sono contenuti all'interno di cisterne limitate da aracnoide e contenenti liquor.
La regione sovrasellare è generalmente approcciata attraverso le cisterne che circondano la parte anteriore dell'incisura tentoriale, definita come uno spazio triangolare situato fra i margini liberi del tentorio. Il chiasma ed i nervi ottici attraversano lo spazio incisurale anteriore, i nervi ottici entrano nel cranio dai canali ottici medialmente alle clinoidi anteriori ed alle carotidi e sono diretti medialmente, posteriormente e superiormente verso il chiasma. Il chiasma è normalmente situato al di sopra del diaframma sellare.
Vista dall'alto la parete laterale del seno cavernoso si estende dalla fessura orbitaria superiore anteriormente, fino all'apice della porzione petrosa dell'osso temporale, posteriormente. Il III nervo cranico entra nel seno dal tetto, lateralmente al dorso sellare, il IV entra in posizione più posteriore e laterale, la branca oftalmica del V entra dalla parte inferiore della parete laterale e il VI entra dalla parete posteriore del seno fra carotide medialmente e III lateralmente.
Una volta entrate nel cranio inferiormente e poi lateralmente al nervo ottico, le carotidi si dirigono lateralmente e danno origine nell'ordine all'arteria oftalmica, alla comunicante posteriore, alla corioidea anteriore, per poi biforcarsi in cerebrale media e cerebrale anteriore, connessa alla cerebrale anteriore controlaterale dalla comunicante anteriore. Dalla carotide nasce l'arteria ipofisaria superiore, che raggiunge il tuber cinereum e si connette alla sua omologa controlaterale per formare un anello attorno all'infundibolo, cui è attaccata l'ipofisi tramite il peduncolo. Gli spazi subaracnoidei della regione sellare e parasellare sono poi attraversati da vasi perforanti che irrorano fra l'altro i nervi ottici, il chiasma, i tratti ottici, le pareti del III ventricolo e l'ipotalamo. Le vene della regione sellare e sovrasellare sono di piccolo calibro e la regione sovrasellare è quasi completamente drenata da tributarie delle vene basali.
Il III ventricolo è situato al centro della testa, superiormente alla sella ed è in stretto rapporto con il poligono di Willis e con il sistema venoso profondo del cervello. La manipolazione delle pareti del III ventricolo può causare disturbi ipotalamici (coscienza, termoregolazione, respirazione, secrezione ormonale, memoria...). E' delimitato da un pavimento, da un tetto, da pareti anteriori, posteriori e laterali. Visto dal basso il pavimento si estende dal chiasma all'acquedotto di Silvio, la metà anteriore è formata da strutture diencefaliche, la metà posteriore da strutture mesencefaliche. Dall'avanti verso l'indietro si trovano: il chiasma, l'infundibolo dell' ipotalamo, il tuber cinereum, i corpi mammilllari, la sostanza perforata posteriore e il tegmento del mesencefalo. L'infundibolo è una struttura imbutiforme localizzata fra il chiasma ed il tuber cinereum. L'ipofisi è connessa all'infundibolo e gli assoni infundibolari si estendono fino al lobo posteriore dell'ipofisi. Il tuber cinereum si fonde nell'infundibolo. La parete anteriore del III ventricolo si estende dai forami di Monro al chiasma inferiormente. L'unica parte visibile è la lamina terminale, un fine strato di sostanza grigia e pia, attaccata alla superficie superiore del chiasma e che riempie il gap fra chiasma e rostro del corpo calloso.
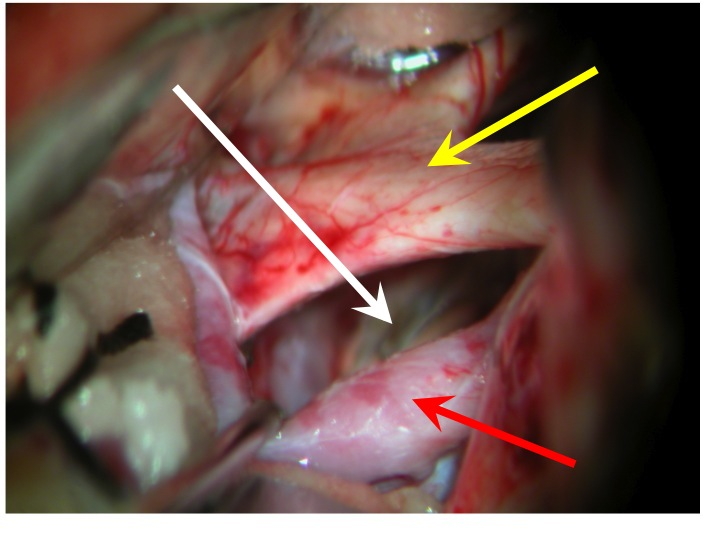
Vista della regione sovrasellare (via pterionale dx).
La freccia rossa indica la carotide dx, la gialla il nervo ottico dx e il chiasma, la bianca il diaframma sellare.
Anatomia dell'ipofisi
L’ipofisi è costituita da un lobo anteriore (adeno-ipofisi), che avvolge la parte più distale del peduncolo ipofisario, costituendo la pars tuberalis, e da un lobo posteriore (neuro-ipofisi), più aderente all'osso della sella di quanto non sia il lobo anteriore. Poichè il lobo anteriore è separato dal lobo posteriore, la pars tuberalis è più frequentemente inserita nel lobo posteriore. Cisti della pars intermedia sono di frequente riscontro.
Nell’adeno-ipofisi sono presenti 6 tipi cellulari diversi:
- le cellule tireotrope, che secernono il TSH;
- le cellule corticotrope, che secernono l’ACTH;
- le cellule lattotrope, che secernono la PRL;
- le cellule somatotrope, che secernono il GH;
- le cellule gonadotrope, che secernono le gonadotropine;
- le cellule follicolo-stellate, che potrebbero rappresentare cellule staminali ipofisarie e la cui funzione sembra importante per la secrezione di fattori di crescita e citochine e per mantenere i corretti rapporti (e quindi l’equilibrio paracrino) fra i diversi tipi cellulari.
Le cellule corticotrope e tireotrope tendono a raggrupparsi insieme nelle zone più centrali della ghiandola, mentre le cellule somatotrope si distribuiscono nelle porzioni più laterali e le cellule gonadotrope, lattotrope e follicolo-stellate sono diffusamente sparse nel parenchima adeno-ipofisario.
La neuro-ipofisi è costituita dalla parte terminale degli assoni delle cellule che secernono vasopressina e ossitocina, i cui corpi cellulari sono contenuti nell’ipotalamo (nei nuclei sopra-ottico e para-ventricolare).
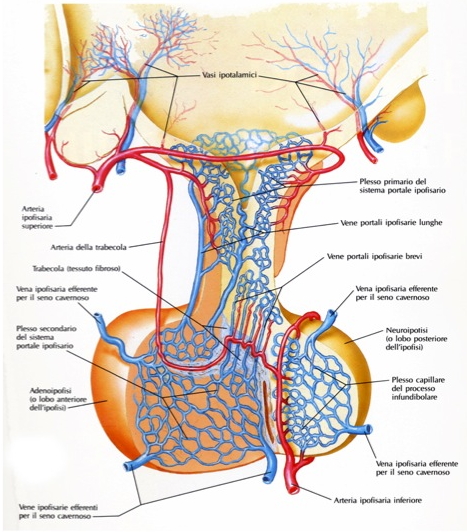
L’ipofisi è vascolarizzata dalle arterie ipofisarie superiori e inferiori e da rami delle arterie comunicanti posteriori, tutte originanti dalla carotide interna o da suoi rami. Le arterie ipofisarie superiori avvolgono la porzione superiore del peduncolo ipofisario, al cui interno comunicano con un plesso primario di capillari sinusoidali spiralizzati. Tali ramuscoli si tramutano in venule e quindi in vene lunghe e sottili che passano sotto il peduncolo ipofisario nella pars distalis dell’adeno-ipofisi, dove si forma un plesso secondario di capillari sinusoidali. Questa disposizione, doppia e parallela, dei plessi capillari prende il nome di sistema portale ipofisario. Il drenaggio venoso fa capo alle vene ipofisarie laterali che si aprono nel seno cavernoso.
Diagnostica generale dell'ipofisi
Generalità di diagnostica biochimica e ormonale ipotalamo-ipofisaria
Imaging neuroradiologico ipotalamo-ipofisario
Campimetria ottica e potenziali evocati visivi