Croniche
Scheda IGF-I ricombinante

Giorgio Radetti & Silvia Longhi
Reparto di Pediatria, Ospedale Regionale di Bolzano
Meccanismo d’azione
IGF-I ricombinante che si lega al recettore naturale.
Indicazioni
Pazienti con sindrome di Laron, casi di mancata produzione o resistenza all’IGF-I.
Controindicazioni
Neoplasia attiva.
Non è raccomandato nei bambini di età < 2 anni per mancanza di dati su sicurezza ed efficacia.
Preparati e dosaggi
Mecasermina (Increlex) è disponibile in fl da 4 mL, contenenti 10 mg/mL (40 mg/fl), da conservare in frigorifero.
Va somministrato per iniezione sottocutanea 2 volte al giorno prima dei pasti. La dose raccomandata iniziale è di 0.04 mg/kg per due volte al giorno, da aumentare gradualmente fino a raggiungere la dose massima di 0.12 mg/kg x 2/die.
Effetti collaterali e precauzioni
Gli effetti collaterali più comuni sono rappresentati dall’ipoglicemia, per cui viene consigliato di somministrare il farmaco prima dei pasti e di eseguire monitoraggio glicemico pre- e post-prandiale, soprattutto all’inizio della terapia.
Occasionalmente è stata descritta ipertrofia nel sito di iniezione e ipertrofia tonsillare. A causa della possibilità di ipertrofia del tessuto linfoide, si consiglia una valutazione otorinolaringoiatrica nel caso insorgano apnea nel sonno, otiti sierose croniche dell’orecchio medio, russamento, ecc.
Un esame del fondo dell’occhio deve essere eseguito qualora compaiano sintomi clinici ricollegabili a ipertensione endocranica, quali cefalea persistente, nausea o vomito (pseudotumor cerebri).
Nei pazienti che manifestano rapida crescita possono verificarsi epifisiolisi e progressione della scoliosi.
Limitazioni prescrittive
Attualmente il farmaco non prevede piano terapeutico nè nota AIFA, ma è un farmaco ospedaliero e per tale motivo è vendibile al pubblico su prescrizione di centri ospedalieri o di specialisti secondo art. 93 DL 219/06.
Scheda GnRH agonisti
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
Meccanismo d’azione
Il GnRH è un decapeptide sintetizzato da un gruppo specializzato di circa 1000 neuroni localizzati nell’ipotalamo. Viene rilasciato in maniera ritmica, con dei pulse che si succedono ogni 30-120 minuti e ai quali corrisponde un’altrettanto ritmica secrezione di LH.
La particolare struttura spaziale ha lo scopo di preservare la posizione di alcuni aminoacidi localizzati in particolari punti chiave, fondamentali per la modulazione dell’affinità recettoriale. La sostituzione della glicina in posizione 6 con diversi altri aminoacidi ha dato vita ad altri peptidi dotati di maggiore affinità recettoriale e maggiore resistenza agli enzimi proteolitici rispetto al GnRH nativo (figura 1) (1).
Dopo un’iniziale e transitoria iperstimolazione, nota come “flare up”, la prolungata esposizione agli analoghi induce una progressiva riduzione della secrezione delle gonadotropine, indotta da diversi meccanismi verosimilmente rappresentati da un’alterazione della trasmissione del messaggio intra-cellulare e/o della sintesi ed esocitosi delle gonadotropine stesse. L’effetto finale è l’inibizione dell’attività gonadica.

Indicazioni
Trattamento della pubertà precoce centrale (2,3), in cui rappresentano il trattamento di scelta dall’inizio degli anni ’80.
Trattamento adiuvante nel cancro della mammella. Nelle donne in pre-menopausa affette da cancro della mammella, l’uso degli analoghi associato ad altri farmaci (anti-estrogeni) sembra ridurre il rischio di recidiva di malattia (4,5).
Trattamento del carcinoma prostatico. Il cancro della prostata, che rappresenta nel mondo la causa principale di morte per la popolazione maschile, ha patogenesi complessa, ma appare evidente una relazione con gli ormoni sessuali (6). Per tale ragione la castrazione o la privazione farmacologica degli androgeni appaiono momenti fondamentali della terapia (7). Gli analoghi più comunemente utilizzati sono leuprolide e goserelin.
Trattamento del dolore da endometriosi. È una patologia estrogeno-dipendente e per questa ragione il trattamento con analoghi è in grado di migliorare il dolore associato a questa condizione (8,9).
Trattamento leiomiomatosi uterina. I leiomiomi rappresentano il tumore benigno più comune nelle donne in età fertile. I sintomi più frequenti sono sanguinamento mestruale eccessivo con relativa anemia, dolore e sensazione di peso nella pelvi e in addome, dolore o fastidio durante i rapporti sessuali, infertilità e aborti ricorrenti (10,11). Gli analoghi del GnRH si sono dimostrati efficaci nel ridurre il volume dei leiomiomi e dell’utero, ma l’effetto è limitato al periodo di trattamento. Sembrano inoltre migliorare l’outcome chirurgico delle donne sottoposte ad isterectomia (12).
Protocolli di procreazione medicalmente assistita, in cui vengono utilizzati da molti anni per prevenire il picco endogeno di LH e scegliere il momento migliore per il prelievo dell’ovocita (13,14).
Prevenzione della menopausa precoce (POF) nelle pazienti oncologiche. La tossicità ovarica è un possibile effetto della chemioterapia e si può manifestare con infertilità, amenorrea transitoria o menopausa precoce. La soppressione dell’attività ovarica indotta dagli analoghi del GnRH potrebbe, attraverso diversi meccanismi, preservare la fertilità della paziente. In letteratura sono presenti diversi lavori che sembrano suggerire con l’uso degli analoghi un minore rischio di POF nelle donne in pre-menopausa sottoposte a chemioterapia, tuttavia al momento il loro utilizzo è controverso, non codificato e quindi off-label (15-17).
Preparazioni, via di somministrazione, posologia
Esistono diverse formulazioni (18):
- forme somministrabili per via nasale o sottocute (tabella 1), che necessitano più somministrazioni giornaliere;
- forme a lento rilascio intra-muscolo o sotto-cute (tabella 2), che assicurano una liberazione costante per 28 giorni o tre mesi. Tra queste certamente quella più utilizzata è la formulazione a somministrazione mensile della leuprolide acetato (19-21);
- impianto sottocute di istrelina in grado di rilasciare il farmaco per un anno (22). L’efficacia sembra analoga a quella ottenuta con la somministrazione di leuprolide, con il grande vantaggio di un lungo intervallo di somministrazione (23,24).
| Tabella 1 Formulazioni short-acting di analoghi del GnRH |
|
| Preparato | Dosaggio/die |
| Nafarelin | spray 800 µg x 2 |
| Buserelin | spray 20-40 µg/kg 1200-1800 µg sc |
| Leuprolide | 50 µg/kg sc |
| Triptorelina | 20-40 µg/kg sc |
| Deslorelina | 4-8 µg/kg sc |
| Istrelina | 8-10 µg/kg sc |
| Tabella 2 Formulazioni long-acting di analoghi del GnRH |
|
| Preparato | Dosaggio |
| Goserelin | 3.6 mg/mese o 10.8 mg/3 mesi |
| Buserelin | 6.3 mg/2 mesi |
| Leuprolide | 3.75 mg/mese o 11.25 mg/3 mesi |
| Triptorelina | 3 o 3.75 mg/mese o 11.25 mg/3 mesi |
| Istrelina | 50 mg impianto annuale |
Tutti i diversi analoghi disponibili sembrano avere analoga efficacia nel trattamento della pubertà precoce centrale (18).
- Leuprolide:
- impianto: 3.6 mg (Leptoprol), 5 mg (Leptoprol),
- siringa preriempita 3.75 mg (Enantone, Politrate), 7.5 mg (Eligard), 11.25 mg (Enantone), 22.5 mg (Eligard, Politrate), 45 mg (Eligard). Nel bambino si usano le fl (intera se > 20 kg, ½ se < 20 kg) da 3.75 mg mensile o da 11.25 mg trimestrale.
- Triptorelina: soluzione 0.1 mg/mL (Decapeptyl, Fertipeptil), 3.75 mg/2 mL (Decapeptyl, Gonapeptyl depot), 11.25 mg/2 mL (Decapeptyl), 22.5 mg/2 mL (Decapeptyl). Nel bambino si usano le fl (½ se < 20 kg, 2/3 tra 20 e 30 kg, intera se > 30 kg,) da 3.75 mg mensile o da 11.25 mg trimestrale.
- Goserelin: siringa preriempita da 3.6 mg (Zoladex), 10.8 mg (Zoladex)
- Buserelin (Suprefact): spray nasale 0.1 mg/puff, soluzione 1 mg/mL, siringa preriempita 6.3 mg, siringa preriempita 9.45 mg
Contro-indicazioni
Gravidanza e allattamento.
Effetti collaterali
Sono collegati alla riduzione della sintesi degli ormoni sessuali, caratteristica propria di questa classe di farmaci.
Più comunemente riportati flushing e cefalea, comunque solitamente di breve durata.
In più del 10% dei pazienti viene riferito dolore nella sede di iniezione, dove raramente sono stati descritti ascessi sterili.
Nei pazienti con carcinoma della prostata, il trattamento induce impotenza, riduzione della libido, osteopenia e disturbi delle vie urinarie (25).
Vampate (più dell’80% delle pazienti), riduzione della libido, secchezza e sanguinamenti vaginali, irritabilità, depressione, riduzione della densità minerale ossea (26).
Limitazioni prescrittive
Nota AIFA 51 per Leuprolide e Triptorelina nella pubertà precoce
Bibliografia
- Millar RP, et al. Current and future applications of GnRH, kisspeptin and neurokinin B analogues. Nat Rev Endocrinol 2013, 9: 451–66.
- Berberoglu M. Precocious puberty and normal variant puberty: definition, etiology, diagnosis and current management. J Clin Res Pediatr Endocrinol 2009, 1: 164–74.
- Lahlou N, et al. Pharmacokinetics and pharmacodynamics of GnRH agonists: clinical implications in pediatrics. J Pediatr Endocrinol Metab 2000, 13 suppl 1: 723–37.
- Cheer SM, et al. Goserelin. A review of its use in the treatment of early breast cancer in premenopausal and perimenopausal women. Drugs 2005, 65: 2639-55.
- Goel S. et al. LHRH agonists for adjuvant therapy of early breast cancer in premenopausal women. Cochrane Database Syst Rev 2009: CD004562.
- Gann PH, et al. Prospective study of sex hormone levels and risk of prostate cancer. J Natl Cancer Inst 1996, 88: 1118–26.
- Shen MM, et al. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010, 24: 1967–2000.
- Kennedy S, et al. ESHRE guideline for the diagnosis and treatment of endometriosis. Hum Reprod 2005, 20: 2698–704.
- Brown J, et al. Endometriosis: an overview of Cochrane Reviews. Cochrane Database Syst Rev 2014: CD009590.
- Evans P, et al. Uterine fibroid tumors: diagnosis and treatment. Am Fam Physician 2007, 75: 1503–8.
- Al-Nafussi A. Uterine smooth-muscle tumours: practical approach to diagnosis. Curr Diagn Pathol 2004, 10: 140–56.
- Stovall TG, et al. GnRH agonist and iron versus placebo and iron in the anemic patient before surgery for leiomyomas: a randomized controlled trial. Leuprolide Acetate Study Group. Obstet Gynecol 1995, 86: 65-71.
- Cetel NS, et al. The dynamics of gonadotropin inhibition in women induced by an antagonistic analog of gonadotropin-releasing hormone. J Clin Endocrinol Metab 1983, 57: 62–5.
- Albano C, et al. Ovarian stimulation with HMG: results of a prospective randomized phase III European study comparing the luteinizing hormone-releasing hormone (LHRH)-antagonist cetrorelix and the LHRH-agonist buserelin. European Cetrorelix Study Group. Hum Reprod 2000, 15: 526–31.
- Chen H, et al. Adjuvant gonadotropin-releasing hormone analogues for the prevention of chemotherapy induced premature ovarian failure in premenopausal women. Cochrane Database Syst Rev 2011: CD008018.
- Partridge A, et al. Age at menopause among women who remain premenopausal following treatment for early breast cancer. Long-term results from International Breast Cancer Study Group Trials V and VI. Eur J Cancer 2007, 43: 1646–53.
- Del Mastro L, et al. Gonadotropin-releasing hormone analogues for the prevention of chemotherapy-induced premature ovarian failure in cancer women: systematic review and meta-analysis of randomized trials. Cancer Treat Rev 2014, 40: 675–83.
- Carel JC, et al. Consensus Statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics 2009, 123: e752-62.
- Lee PA, et al. Efficacy and safety of leuprolide acetate 3-month depot 11.25 milligrams or 30 milligrams for the treatment of central precocious puberty. J Clin Endocrinol Metab 2012, 97: 1572–80.
- Lee PA, et al. Efficacy of leuprolide acetate 1-month depot for central precocious puberty (CPP): growth outcomes during a prospective, longitudinal study. Int J Pediatr Endocrinol 2011, 2011: 7.
- Neely EK, et al. Leuprolide acetate 1-month depot for central precocious puberty: hormonal suppression and recovery. Int J Pediatr Endocrinol 2010, 2010: 398639.
- Eugster EA, et al. Efficacy and safety of histrelin subdermal implant in children with central precocious puberty: a multicenter trial. J Clin Endocrinol Metab 2007, 92: 1697–704.
- Rahhal S, et al. Results of a second year of therapy with the 12-month histrelin implant for the treatment of central precocious puberty. Int J Pediatr Endocrinol 2009, 2009: 812517.
- Gillis D, et al. Time to menarche and final height after histrelin implant treatment for central precocious puberty. J Pediatr 2013, 163: 532-6.
- Choi S, Lee AK. Efficacy and safety of gonadotropin-releasing hormone agonists used in the treatment of prostate cancer. Drug Healthc Patient Saf 2011, 3: 107–19.
- Magon N. Gonadotropin releasing hormone agonists: Expanding vistas. Indian J Endocrinol Metab 2011, 15: 261-7.
Displasie scheletriche
Tiziana Greggi
Chirurgia delle Deformità del Rachide, Istituto Ortopedico Rizzoli, Bologna
(aggiornato al novembre 2022)
INTRODUZIONE
Le displasie scheletriche sono un vasto (oltre 400 tipi) ed eterogeneo gruppo di anomalie delle modalità di crescita e sviluppo del tessuto osteo-cartilagineo delle ossa, da causa generalmente genetica. Sono note da numerosi decenni, soprattutto in ambiente ortopedico-pediatrico per la necessità di trattamenti chirurgici.
L’espressività clinica varia dalle forme letali in epoca peri-natale, a bassa statura, talvolta disarmonica, a condizioni di lieve ritardo di crescita. La caratterizzazione clinico-radiologica risulta spesso semplice e ben schematizzabile, mentre la definizione pato-molecolare è molto complessa.
La prevalenza varia molto nei diversi studi (da 2.1 a 4.7/10mila nati), ma quella ritenuta più reale è di 2 casi su 10mila nati vivi e 20 casi su 10mila nati morti (1). Le forme più comuni sono l’acondroplasia, l'acondrogenesi, la displasia tanatofora e l’osteogenesi imperfetta.
Per gli aspetti anatomici, istologici e di struttura del tessuto osseo bisogna fare un breve cenno ai due processi di sviluppo del tessuto osseo e ad alcune fasi dell'embriogenesi. Circa alla 5° settimana di gestazione compaiono gli arti superiori e, qualche giorno dopo, gli arti inferiori; alla 7° appare la cartilagine, alla 12° sono presenti i centri di sviluppo delle ossa lunghe (fig 1).
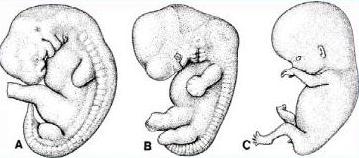
Figura 1
Sviluppo del feto: A = 5.5 settimane; B = 7 settimane; C =12 settimane
L’osso dello scheletro si genera per:
- ossificazione diretta membranosa del mesenchima, a formare le ossa della volta cranica e delle clavicole, in cui le cellule progenitrici mesenchimali si differenziano via pre-osteoblasti in osteoblasti;
- ossificazione indiretta encondrale (cartilaginea) delle ossa lunghe, vertebre e coste, dove le cellule mesenchimali progenitrici si differenziano dapprima in cellule pericondriali e condrociti e successivamente si trasformano in osteoblasti.
Per chiarire lo schema di ossificazione encondrale: alle estremità delle ossa lunghe c’è la cartilagine di accrescimento, dove si distinguono 4 zone cellulari (di riserva, di proliferazione, pre-ipertrofica e ipertrofica), in cui si completa il modello cartilagineo che subirà l’apposizione di tessuto osseo e la sua sostituzione con l’invasione contemporanea dei vasi sanguigni. La crescita in senso latero-laterale è dovuta alle cellule pericondriali, che diverranno cellule del periostio nell’osso maturo (fig 2).
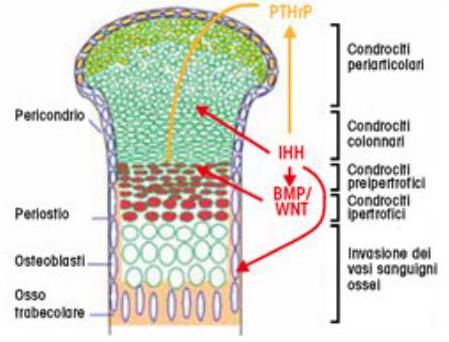
Figura 2
Via di segnale che determina la crescita della cartilagine di accrescimento e la formazione del tessuto osseo. Lo sviluppo della cartilagine di accrescimento è determinato dal controllo di diverse vie di segnale, tra cui IIHH e PTHrP, BMPs, WNT. Tutte inducono lo sviluppo della zona di proliferazione dei condrociti e ne inibiscono la maturazione, contrastando l'azione contraria della via FGFs/FGFr. Lo stampo cartilagineo verrà poi riassorbito dall'azione di metallo-proteasi, sostituito da osteoblasti e vasi sanguigni, quindi da tessuto osseo maturo.
È importante ricordare che il tessuto osseo è dinamico: grazie all’equilibrio funzionale delle due linee cellulari (osteoblasti e osteoclasti), subisce un continuo e regolare processo di riassorbimento e neoformazione (2).
Cercherò qui di presentare sinteticamente questo vastissimo argomento, puntualizzando le principali caratteristiche di queste condizioni patologiche, facendo riferimento alle forme più comuni e alle classificazioni più attuali.
CLASSIFICAZIONE
Ci sono stati numerosi tentativi di portare ordine in questo campo, elaborando una classificazione delle malattie costituzionali dello scheletro, contenente al primo posto il capitolo delle osteo-condro-displasie (3,4). Secondo una recente revisione del 2019 del gruppo di esperti dell’International Skeletal Dysplasias Society, sono classificate 461 condizioni di displasia scheletrica, suddivise in 42 diversi gruppi con specifici criteri clinici, radiografici e/o molecolari (5) (tab 1).
| Tabella 1 Classificazione delle displasie scheletriche genetiche (5) |
||
| # | Gruppo | Esempi |
| 1 | Condro-displasie correlate a FGF-R3 | Acondroplasia, ipocondroplasia |
| 2 | Collagene di tipo 2 | S. di Stickler |
| 3 | Collagene di tipo 11 | Fibrocondrogenesia |
| 4 | Disordini della solfatazione | S. di Ehlers-Danlos |
| 5 | Perlecan | Condrodistrofia miotonica |
| 6 | Aggrecan | Ipostaturismo con età ossea avanzata |
| 7 | Filamina | Displasia fronto-metafisaria |
| 8 | TRPV4 | Displasia metatropica |
| 9 | Ciliopatie | Displasia toracica asfissiante |
| 10 | Displasie epifisarie multiple e pseudo-acondroplasia | Pseudo-acondroplasia |
| 11 | Displasie metafisarie | S. di Shwachman–Bodian–Diamond |
| 12 | Displasie spondilo-metafisarie | Spondilo-encondrodisplasia |
| 13 | Displasie spondilo-epi-(meta)-fisarie | Displasia di Dyggve–Melchior–Clausen |
| 14 | Displasie spondilo-displasiche severe | Acondrogenesia |
| 15 | Displasie acromeliche | Acrodisostosi |
| 16 | Displasie acro-mesomeliche | Displasia acro-mesomeliche di Maroteaux |
| 17 | Displasie mesomeliche e rizo-mesomeliche | Discondroplasia di Leri-Weill |
| 18 | Displasie con incurvamento osseo | Displasia camptomelica |
| 19 | Nanismi primitivi con ossa sottili | S. di Kenny-Caffey, s. IMAGE |
| 20 | Displasie con dislocazioni articolari multiple | Displasia di Desbuquois |
| 21 | Condrodisplasia puntata | S. di Keutel |
| 22 | Displasie osteosclerotiche neonatali | Displasia di Blomstrand |
| 23 | Osteopetrosi | Osteopetrosi neonatale severa, picnodisostosi |
| 24 | Disordini osteosclerosanti | Displasia diafisaria di Camurati-Engelman |
| 25 | Osteogenesi imperfetta | Osteogenesi imperfetta tipi 1-6 |
| 26 | Anomalie della mineralizzazione | Ipofosfatasia, rachitismi ipofosfatemici |
| 27 | Malattie da accumulo lisosomiale con coinvolgimento osseo | Mucopolisaccaridosi |
| 28 | Malattie osteolitiche | Progeria di Hutchinson-Gilford |
| 29 | Sviluppo disorganizzato delle componenti scheletriche | S. di McCune-Albright, neurofibromatosi tipo 1, fibrodisplasia ossificante progressiva |
| 30 | Alta statura con coinvolgimento scheletrico | S. di Sotos, s. di Marfan |
| 31 | Osteoartropatie genetiche infiammatorie simil-reumatiche | Displasia pseudo-reumatoide progressiva |
| 32 | Displasie cleido-craniche | Displasia cleido-cranica |
| 33 | Cranio-sinostosi | S. di Crouzon |
| 34 | Disostosi con coinvolgimento cranio-facciale predominante | S. di Treacher-Collins |
| 35 | Disostosi con predominante coinvolgimento vertebrale (con e senza coinvolgimento costale) | S. di Klippel-Feil |
| 36 | Disostosi rotulee | S. della rotula piccola |
| 37 | Brachidattilie (senza manifestazioni extra-scheletriche) | S. di Cooks |
| 38 | Brachidattilie (con manifestazioni extra-scheletriche) | Pseudoipoparatiroidismo tipo IA |
| 39 | Ipoplasie degli arti | Sirenomelia |
| 40 | Ectrodattilia | S. di Hartsfield |
| 41 | Polidattilia-sindattilia-trifalangismo | S. di Pallister-Hall |
| 42 | Difetti della formazione articolare e sinostosi | S. di Liebenberg |
CONSIDERAZIONI GENERALI CLINICO-DIAGNOSTICHE E DI TRATTAMENTO
Ciascun tipo di displasia scheletrica è caratterizzato da un insieme diverso di anomalie.
Gli aspetti clinici comuni fondamentalmente sono: bassa statura, braccia e gambe corte, dita corte, testa sproporzionatamente grande, collo corto, mobilità ridotta all’altezza dei gomiti. Alcuni tipi di displasia scheletrica vengono rilevati a circa 20 settimane di gravidanza nel corso di un’ecografia, mentre altri possono non risultare evidenti fino alla prima infanzia. Anche se la displasia scheletrica viene rilevata nel corso della gravidanza, può risultare difficile diagnosticarne il tipo esatto prima del parto, soprattutto per la difficoltà di approfondimenti diagnostico-strumentali, meglio eseguibili quando i problemi della crescita si presentano in età pediatrica o nella prima infanzia (6,7).
Per quel che riguarda la prognosi, non c'è sopravvivenza quando la displasia scheletrica causa crescita ossea particolarmente anomala, che impedisce al torace e ai polmoni di svilupparsi in modo corretto. Tale condizione caratterizza le displasie scheletriche letali e si verifica in circa 1 neonato su 10mila. Si definisce invece non letale la displasia scheletrica in cui i neonati colpiti sopravvivono al parto e al periodo immediatamente successivo. Tra i bambini affetti da displasia scheletrica non letale, le diagnosi più comuni sono l’acondroplasia, spesso indicata con il nome di nanismo, e l'osteogenesi imperfetta detta anche "malattia delle sclere blu".
Il sospetto diagnostico viene posto inizialmente tramite la raccolta dell’anamnesi familiare e personale del bambino e l’esame obiettivo, con particolare attenzione alla valutazione auxologica e alla ricerca di alcuni dismorfismi caratteristici. In particolare, la valutazione clinica deve comprendere la rilevazione dei seguenti parametri: peso, lunghezza e circonferenza cranica alla nascita, valutazione della curva di crescita, sia staturale sia ponderale, e della curva di aumento della circonferenza cranica, rapporto tra segmento corporeo superiore e inferiore, descrizione dell’eventuale accorciamento delle estremità, caratteristiche del cranio, anomalie dentali, scoliosi, lordosi, varismo o valgismo del ginocchio. Dovrebbero essere valutate inoltre le caratteristiche di pelle, unghie e capelli, insieme a disturbi cognitivi e potenziali anomalie dell’udito e/o della vista.
La conferma diagnostica si ottiene tramite test genetici specifici. Dovrebbero essere eseguiti esami ematici generali, oltre a test renali, epatici e cardiaci per valutare l’eventuale presenza di complicanze a carico di tali organi. È necessaria una valutazione radiologica per definire la gravità della patologia e l’eventuale presenza di complicanze (8,9).
Il trattamento delle displasie scheletriche è multi-disciplinare. La gestione del bambino può essere ottimizzata utilizzando un team specialistico che dovrebbe coinvolgere pediatri, genetisti, neurologi e neurochirurghi pediatrici, pneumologi pediatrici e chirurghi ortopedici pediatrici.
L’assistenza preventiva è essenziale e ogni sforzo deve essere diretto a identificare i bambini ad alto rischio di sviluppare complicanze, in modo da mettere in atto interventi terapeutici appropriati e precoci, per prevenire sequele permanenti.
Negli ultimi anni si sono delineate nuove promettenti prospettive terapeutiche, mirate ai meccanismi alla base della malattia e non solo alla correzione chirurgica dei difetti ad essa associati. L’intervento chirurgico è una forma comune di terapia per l’ipostaturismo (nanismo), sia armonico sia disarmonico. Tuttavia, l’indicazione chirurgica rimane controversa, poiché si tratta di una procedura dolorosa e associata a complicanze, che includono infezioni, contratture muscolari e aumento del rischio di fratture (10,11). L’allungamento dell’arto attraverso la procedura di Ilizarov comporta, infatti, la rottura chirurgica delle ossa lunghe, la fissazione esterna e la distrazione graduale per molti mesi durante il processo di guarigione. La lunghezza media ottenuta è di circa 20.5 cm dopo multiple procedure applicate sia al femore sia alla tibia. È necessaria un’attenta valutazione clinica e psicologica pre-operatoria, per valutare l’alto rischio di complicanze rispetto al miglioramento staturale ottenibile. Le recenti innovazioni, come l’uso della fissazione intra-midollare, possono migliorare i risultati e ridurre i rischi (12-14). In futuro, la combinazione dell’allungamento chirurgico dell’arto con le strategie farmacologiche potrebbe condurre a un ulteriore miglioramento della prognosi staturale di questi pazienti.
Attualmente non esistono terapie farmacologiche approvate per le condro-displasie, ad eccezione dell’ormone della crescita, che ha indicazione per l’acondroplasia soltanto in Giappone. Negli ultimi anni sono state proposte e studiate molte strategie non chirurgiche, sfruttando l'impiego di anticorpi monoclonali volti a ridurre l’eccessiva attivazione di FGF-R3, in modo da stimolare la crescita ossea lineare nei pazienti affetti da acondrodisplasia, o di recente la disponibilità di un anticorpo monoclonale, il burosumab, per i pazienti con la forma di rachitismo ipofosfatemico X-linked (11,15,16). Nonostante ci troviamo già in fase di sperimentazione di numerose nuove promettenti terapie farmacologiche (11,16-18), oggi per cercare di migliorare la qualità e la durata della vita di chi è affetto da displasia scheletrica bisogna spesso considerare l'impiego di soluzioni chirurgiche. È tuttora attuale la necessità di correggere chirurgicamente la direzione in cui crescono le ossa o per allungare gli arti, per eseguire osteo-sintesi di fratture ripetute, per correggere la colonna vertebrale, per allargare il canale vertebrale, per inserire uno shunt atto a drenare l’ipertensione endo-cranica.
I trattamenti riabilitativi fisiatrici sono di estrema necessità per rafforzare i muscoli e aumentare l’estensione dei movimenti. Per il trattamento di questi stati patologici è indispensabile la messa a punto e personalizzazione di attrezzature di assistenza e modi alternativi per lo svolgimento delle attività quotidiane. I chirurghi ortopedici pediatrici, i neurochirurghi e i fisiatri non sono gli unici operatori, ma agiscono in collaborazione con genetisti, cardiologi, otorinolaringoiatri, oftalmologi, neurologi, neuropsichiatri infantili, endocrinologi, fisiatri, avvalendosi inoltre di un adeguato counseling e supporto psicologico.
SISTEMATICA DELLE DISPLASIE SCHELETRICHE: EZIOLOGIA E DIAGNOSI
Queste patologie possono derivare da difetti genetici, noxae esogene o da sindrome della banda amniotica.
Evitiamo di affrontare la classificazione molecolare molto estesa e accurata, basata sulle diverse alterazioni genetiche presenti nelle displasie scheletriche, che agiscono sulla cartilagine di accrescimento, limitandoci a segnalare che possono essere anomalie genetiche che agiscono nello sviluppo della cartilagine di accrescimento o coinvolgono la sintesi di proteine extra-cellulari (tab 2-4).
| Tabella 2 Anomalie genetiche che agiscono nello sviluppo della cartilagine di accrescimento |
|||
| Condizione | Geni | Trasmissione | Segni clinici e radiologici |
| Segnale FGF | |||
| Acondroplasia | FGF-R3 | Autosomica dominante (AD) | Macrocrania, ipoplasia media facies, bozze frontali prominenti. Riduzione rizomelica (parte prossimale) degli arti, limitata estensione del gomito, mano a tridente, ginocchio varo, iperlordosi lombare. Ossa lunghe corte e ricurve, pelvi a tridente, ridotta distanza fra i peduncoli vertebrali lombari. |
| Displasia tanatofora tipo I e II | FGF-R3 | AD | Forma letale nel periodo peri-natale, arti molto corti, femori a cornetta del telefono, marcata plati-spondilia e ristrettezza del torace, cranio a trifoglio (tipo II). |
| Ipocondroplasia | FGF-R3 | AD | Ritardo di crescita pre- e post-natale, intelligenza nella norma. Ipoplasia rotulea, iperlassità articolare. Enfisema, tracheo-laringo-bronco-malacia. Difficoltà respiratorie e nell'alimentazione nel primo anno di vita. Anomalie genitali, ipoplasia mammaria. |
| Segnale BMP | |||
| Brachidattilia A1, A2, C | IHH, GDF5, BMPR1B, BMP2, CDMP1 | AD | Anomalie di tutte le falangi medie e fusione con le distali (A1). Anomalie della falange media dell'indice e del secondo dito del piede (A2). Anomalie falangi medie II e III dito mano, anomalie solo falange media V dito, II e IV dito più lungo. |
| Segnale WNT | |||
| Sindrome di Robinow | WNT5A DWL1 DWL3 |
AD RS1 AD RS2 AD RS3 |
Esordio post-natale, bassa statura, brevità acro-mesomelica degli arti, macrocefalia, bozze frontali, occhi prominenti, ipertelorismo, narici antiverse. Appiattimento della regione media della facies. Anomalie dentali (malocclusione, ipodonzia, ritardo eruzione denti permanenti, denti sovrannumerari).
|
| ROR2 | Autosomica recessiva (AR) | Possibili anomalie cardiache e renali, labio-palatoschisi, displasie ungueali. Riduzione mesomelica soprattutto degli arti superiori, brachidattilia. Segmentazione vertebrale. Una variante allelica AD ha brachidattilia con falangi medie corte, falangi distali rudimentali o assenti, deformità del pollice, alluci grandi. |
|
| Segnale PTHrO | |||
| Displasia acro-capito-femorale | IHH | AR | Bassa statura disarmonica, macrocrania relativa, brachidattilia, epifisi a cono alle mani e femore. La forma allelica AD (causata non solo da IHH, ma anche da GDF5 e BMPR1B) presenta brachidattilia tipo A1, tipica per le anomalie di tutte le falangi medie fuse con le distali. |
| Displasia metafisaria tipo Jansen | PTH-R1 | AD | Bassa statura disarmonica grave, faccia prominente con mandibola piccola, arti corti e curvi. Ipercalcemia e iposfatemia. |
| Condrodisplasia tipo Blornstrand | PTH-R1 | AD | Displasia letale, polidramnios, idrope fetale. Anomalie facciali, arti molto corti. Incremento della densità ossea, con maturazione scheletrica avanzata. |
| Segnale CNP/NPR2 | |||
|
Displasia acromesomelica tipo Maroteaux |
NPR2 | AR | Bassa statura disarmonica, con riduzione dei segmenti mesomelici ed acromelici dei quattro arti. Fronte prominente, con naso piccolo e largo. Pectus escavatum/carenato, ipercifosi dorsale, iperlordosi lombare. Intelligenza normale. |
| Bassa statura idiopatica armonica | NPR2 | AD | Bassa statura moderata. Riduzione segmenti mesomelici arti superiori ed inferiori. |
| Tabella 3 Mutazioni genetiche con ruolo non ancora ben definito nella replicazione del DNA |
|||
| Condizione | Geni | Trasmissione | Segni clinici e radiologici |
| Sindrome di Kenny-Caffey | FAM11A | AD | Importante bassa statura, anomalie facciali e oculari, mani e piedi piccoli. Ispessimento della corticale delle ossa lunghe. Stenosi midollare, cranio-stenosi, ritardo chiusura fontanella anteriore. Ipoparatiroidismo, ipocalcemia, possibile ipofosfatemia e anemia. Calcificazioni renali e dei nuclei della base. Possibile epilessia. Intelligenza normale, voce con timbro acuto. |
| Discondrosteosi (sindrome di Léri-Weill) | SH0X | Aplo-insufficienza | Bassa statura mesomelica (brevità degli arti nella porzione media), anomalia di Madelung (curvatura dell'avambraccio per disallineamento fra radio, ulna e ossa carpali). Intelligenza nella norma. |
| Displasia mesomelica di Langer | SH0X | Aplo-insufficienza | Bassa statura severa, brevità delle ossa lunghe. Ipoplasia ulna e perone. Anomalia di Madelung assente. Intelligenza normale. |
| Tabella 4 Difetti delle proteine della matrice extra-cellulare |
|||
| Condizione | Geni | Trasmissione | Segni clinici e radiologici |
| Aggrecano | |||
| Displasia spondilo-epi-metafisaria tipo aggrecano | ACAN | AR | Bassa statura armonica nell'infanzia, disarmonica nell'età adulta. Macrocefalia relativa. Marcata ipoplasia della regione centrale della faccia, con assenza della cartilagine nasale, prognatismo. Collo corto. Torace a botte. Platispondilia e schisi dei corpi vertebrali cervicali. Scoliosi e iperlordosi lombare. Riduzione rizo-mesomelica degli arti, con epifisi irregolari e metafisi allargate. Brachidattilia mani, con pollici larghi. |
| Displasia spondilo-epifisaria tipo Kimberley | ACAN | AD | Habitus tarchiato, con bassa statura armonica. Irregolarità delle cartilagini di accrescimento. Sclerosi dei corpi vertebrali e anomalie epifisarie con ritardo dell'età ossea. Artropatia progressiva severa e precoce. |
| Osteocondrite dissecante familiare | ACAN | AD | Bassa statura disarmonica. Riduzione degli spazi inter-vertebrali. Frammentazione della cartilagine articolare e dell'osso subcondrale, con frammenti liberi intra-articolari, versamento articolare con articolazione a scatto. |
| Bassa statura con età ossea avanzata | ACAN | AD | Età ossea avanzata con blocco anticipato della crescita staturale. |
| Collagene tipo 1 | |||
| Osteogenesi imperfetta | COL1A1 | AD |
Tipo I lieve: fragilità ossea, sclere blu, ipoacusia. Raramente bassa statura e dentinogenesi imperfetta. |
| IFITM5 | AD | Tipo V moderata: fragilità ossea, iperplasia del callo osseo, calcificazione della membrana inter-ossea, assenza di sclere blu e dentinogenesi imperfetta. | |
| Collagene tipo 2 | |||
| Displasia spondilo-epifisaria congenita | COL2A1 | AD |
Bassa statura disarmonica, con prevalenza di tronco corto. Facies piatta, ponte nasale sottile, talora palatoschisi, miopia. Collo corto, petto carenato, ginocchio valgo. Quadro radiologico diverso in rapporto all'età:
|
| Displasia di Kneist | COL2A1 | AD | Faccia mediana piatta, radice nasale infossata, palatoschisi, miopia. Tronco corto, iperlordosi lombare, scoliosi. Arti corti con articolazioni prominenti. Segni radiologici specifici: plati-spondilia, corpi vertebrali con cuneizzazione anteriore, "cleft" coronale lombare. Anomalie epifisarie ossa lunghe, collo femorale corto e largo. |
| Collagene tipo 9 | |||
| Displasia epifisaria multipla |
COL9A1 |
AD |
Bassa statura disarmonica con arti corti. Dolore e tumefazione articolare. Osteo-artrite a esordio precoce. Deambulazione dondolante (anserina). Coinvolgimento di tutte le epifisi, in particolare anca, ginocchio, caviglia, mani e polsi. Appiattimento delle epifisi con l'età, metafisi normali, lieve riduzione di lunghezza ossa lunghe, vertebre con minime anomalie. Segno particolare: rotula bipartita. |
| Pseudo-acondroplasia | COMP | AD | Bassa statura disarmonica. ossa lunghe incurvate. Brachidattilia, con iperlassità soprattutto inter-falangea e carpale. Anomalie vertebrali. Osteo-artrite. Assenza delle caratteristiche facciali proprie dell'acondroplasia. Segni radiologici tipici sono i corpi vertebrali biconvessi, con protrusione anteriore linguiforme, corpi vertebrali normali in adolescenza, anomalie epifisarie diffuse, soprattutto del femore prossimale. |
| Sindrome di Stickler | tipo 1 COL2A1 tipo 2 COL1A1 tipo 3 COL1A2 |
AD |
Displasia ossea spondilo-epimetafisaria. Iperlassità articolare. Osteo-artrite progressiva. Micro-retrognatia, con anomalie del palato, sequenza di Pierre Robin. Ipoacusia. Miopia grave, con distacco di retina ed anomalie del corpo vitreo. |
| COL9A1 COL9A2 COL9A3 |
AR | ||
DIAGNOSI PRE-NATALE
Sono fondamentali: l'anamnesi familiare ed ostetrica, con la datazione corretta della gestazione, unite alla misurazione delle ossa lunghe e delle dimensioni del cranio (circonferenza cranica — HC, circonferenza toracica — CC, circonferenza addominale — AC, lunghezza femorale — FL).
L'esame ecografico nelle diverse fasi della gravidanza permette di:
- I trimestre: identificare gli arti e i diversi segmenti ossei;
- II trimestre: valutare lunghezza, morfologia, motilità, atteggiamento ed ecogenicità dei quattro arti;
- III trimestre: valutare lunghezza degli arti e mineralizzazione ossea.
Nel sospetto di ritardo di crescita intra-uterino (IUGR), misurare il rapporto FL/piede (fig 3).
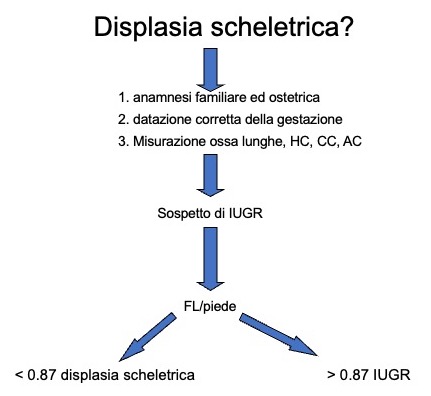
Figura 3
Iter diagnostico per differenziare displasie scheletriche da IUGR
La bassa statura è uno degli elementi clinici per cui più frequentemente un paziente viene sottoposto all’attenzione del pediatra. La Società Europea di Endocrinologia Pediatrica (ESPE) ha proposto nel 2007 una classificazione della bassa statura, aggiornata nel 2016, che la suddivide in tre grandi categorie (5):
- primaria, che comprende le condizioni sindromiche su base genetica;
- secondaria, che comprende i nati piccoli per età gestazionale, con deficit di recupero della crescita;
- idiopatica, che comprende le displasie scheletriche.
Le displasie scheletriche si differenziano come gravità fra:
- forme letali, che si caratterizzano per FL/AC < 0.16, polidramnios, micromelia severa, ipoplasia toracica severa;
- forme non letali, in cui è comunque fondamentale la comparazione con gli aspetti standard dei segmenti ossei, la valutazione qualitativa dell’osso (demineralizzazione, fratture), la valutazione di mani, piedi e cranio (macrocrania, prominenza delle bozze frontali, ipo-ipertelorismo), liquido amniotico, colonna vertebrale, visceri, ecocardiogramma, movimenti fetali.
Il test molecolare e il counseling genetico completano la diagnosi.
DISPLASIE SCHELETRICHE LETALI (0.95-1/10mila nati)
Acondrogenesi
Genetica: deriva da una rara mutazione del gene codificante per il collagene tipo II. Ne esistono due sottotipi, il tipo I (AR) e il tipo II (AD).
Clinica: micromelia estrema.
Diagnosi: ossa appena evidenziabili e ricurve, ipoplasia toracica severa, ipomineralizzazione di grado variabile.
Esito: letale nel 100% dei casi.
Displasia tanatofora
Genetica: deriva da una mutazione relativamente frequente del gene codificante per Fibroblast Growth Factor Receptor.
Clinica: rizomelia severa, ipoplasia toracica, cranio a trifoglio (in una delle varianti).
Esito: letale nel 100% dei casi per ipoplasia toracica.
Osteogenesi imperfetta
Comprende un gruppo eterogeneo di patologie congenite (tab 5), caratterizzate da estrema fragilità ossea, per mutazione dei geni codificanti per il collagene tipo I.
Dopo le prime descrizioni del 1788, sono stati introdotti vari sinonimi: malattia di Lobstein, malattia di Vrolik, osteosatirosi, fragilitas ossium.
| Tabella 5 Classificazione clinica e radiografica dell’osteogenesi imperfetta |
|||
| Tipo | Trasmissione | Statura | Gravità e manifestazioni |
| I | AD | Normale | Lieve: fragilità ossea, sclere blu, ipoacusia (raramente bassa statura e dentinogenesi imperfetta). |
| II | AD | Normale | Letale nel periodo peri-natale, sclere blu, assenza di mineralizzazione, ossa curve con fratture pre-natali, insufficienza respiratoria. |
| III | AD | Bassa | Severa con deformità: scoliosi, spondilo-listesi, schiacciamenti vertebrali, instabilità cervicale C1-C2, invaginazione del dente dell’epistrofeo nella base cranica, con compressione del ponte e manifestazioni neurologiche anche severe (atassia, disfagia), sclere blu, dentinogenesi imperfetta. |
| IV | AD | Bassa | Da moderata a severa fragilità ossea, con fratture frequenti, ma meno numerose alle ossa lunghe, non sclere blu. |
| V | AD | Lievemente diminuita | Fragilità ossea da lieve a moderata, formazione di callo ipertrofico dopo frattura e calcificazione della membrana interossea dell’avambraccio, dentinogenesi imperfetta, assenza di sclere blu. |
| VI | AD | Normale o lievemente diminuita | Alta fragilità ossea con fratture frequenti sin dall’infanzia, fratture vertebrali da compressione, deformità degli arti. Assenza di sclere blu e difetti della dentinogenesi. |
| VII | AR | Variabile | Fratture intra-uterine, deformità degli arti inferiori, accorciamento rizomelico degli arti inferiori, coxa vara. |
Le caratteristiche principali sono 4: difetto genetico, collagenopatia, fragilità ossea, fratture frequenti. Oltre all'osso, sono colpiti altri tessuti, di cui il collagene I è il principale costituente: sclere, dentina, ossa dell’apparato acustico, cute, vasi, capillari, valvole cardiache (fig 4,5).
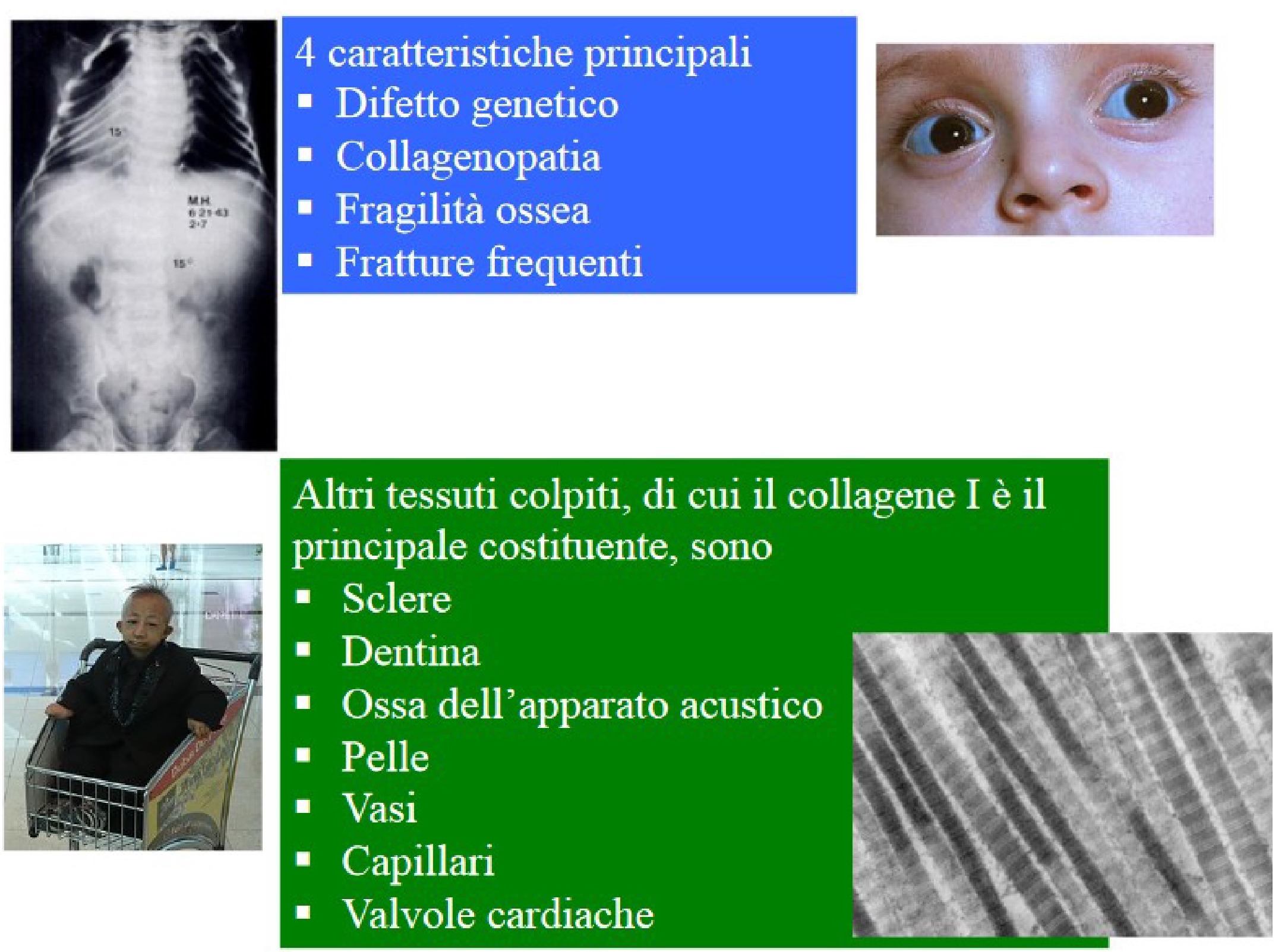
Figura 4
Caratteristiche cliniche dell'osteogenesi imperfetta
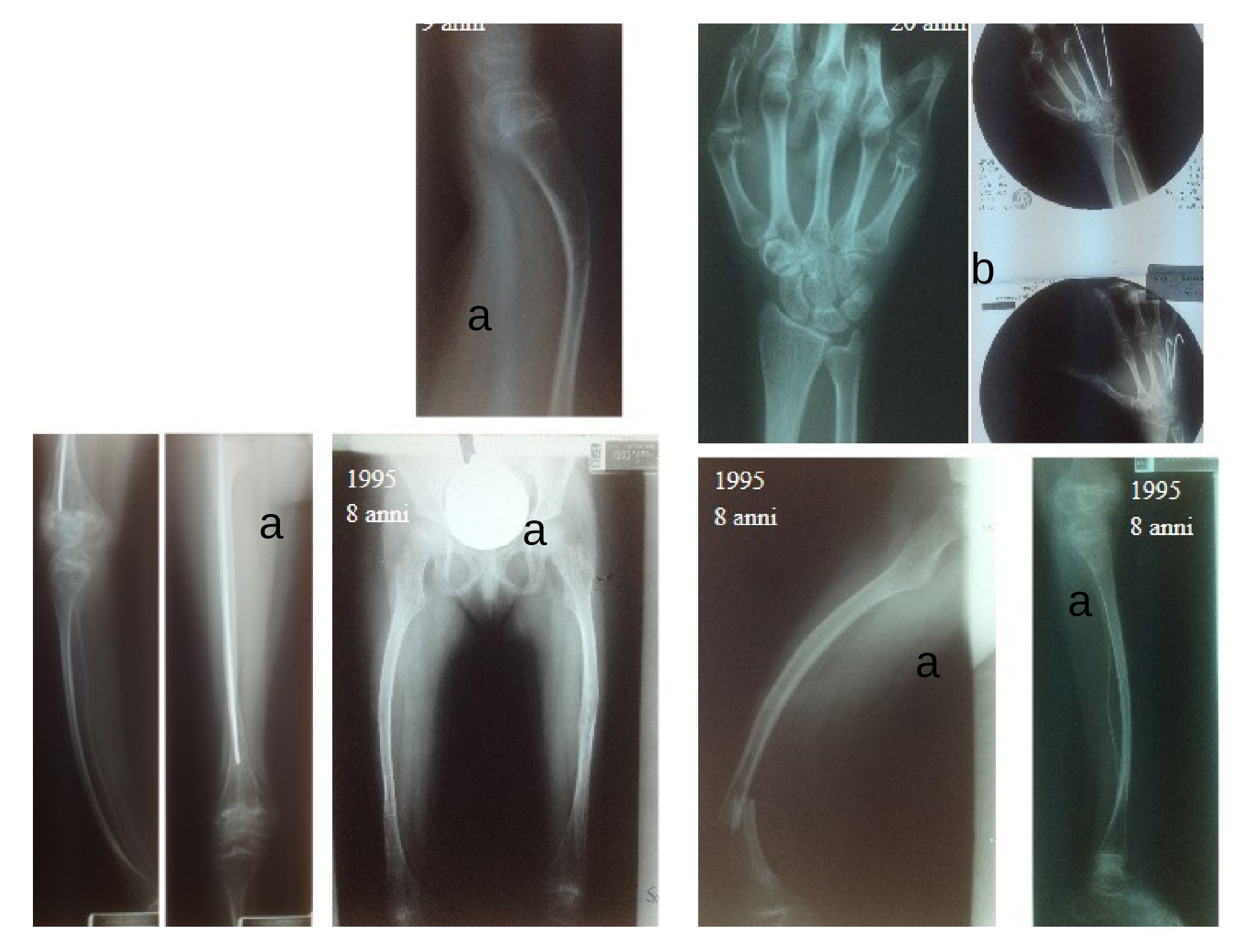
Figura 5
Ragazza con osteogenesi imperfetta di tipo III (trattata chirurgicamente per scoliosi a 15 anni), trattata chirurgicamente più volte in età pediatrica e adulta alle mani e agli arti inferiori.
Incidenza: relativamente frequente (0.4/10mila), di cui la metà è costituita dal tipo II.
Clinica: fratture spontanee, ipoplasia toracica, ipomineralizzazione, ossa deformate (incurvatura). Interessamento molto severo del rachide, con scoliosi dal 30% al 90% dei casi (aumenta con l’aumentare della severità della patologia). L’alta prevalenza di scoliosi è a sostegno della teoria che collega la collagenopatia con iperlassità dei legamenti della colonna e deformità a carico dei corpi vertebrali e frequenti micro-fratture vertebrali da fragilità, al danno a carico delle cartilagini di accrescimento, al verificarsi di fratture e deformità costali, oltre all'impossibilità di deambulare correttamente e ai problemi legati alla dentinogenesi imperfetta. La scoliosi è meno frequente prima dei 6 anni, ma poi va spesso in rapida progressione. Se la progressione della curva scoliotica non viene arrestata tempestivamente, la deformità può evolvere verso valori molto elevati (oltre i 90°) (fig 6), causando riduzione della funzionalità polmonare e sviluppo di cuore polmonare, con esito infausto.
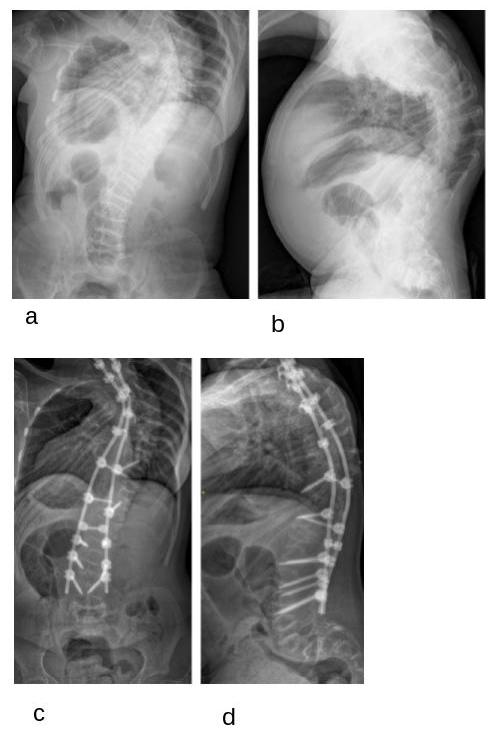
Figura 6
Radiografie in ortostatismo di ragazza di 15 anni con osteogenesi imperfetta di tipo III, con severa scoliosi, precedentemente trattata con bisfosfonati e con ortesi bassa: pre-operatorie antero-posteriore (a) e latero-laterale (b); antero-posteriore (c) e latero-laterale (d), tre mesi dopo correzione ed artrodesi posteriore strumentata mediante viti peduncolari.
Diagnosi: può essere effettuata in fase pre-natale (nei casi gravi), clinicamente, radiograficamente o mediante esame biochimico o genetico.
Trattamento medico: bisfosfonati, anche in pazienti di età < 2 anni (tab 2-5).
Trattamento ortesico (busto ortopedico o corsetto in gesso): è controverso, perchè può causare deformità costali severe, con aggravamento secondario della scoliosi (è sicuramente controindicato un corsetto alto con mentoniera, che può causare malocclusione dentale e piaghe da decubito).
Trattamento chirurgico: oltre al trapianto di midollo osseo per aumentare la densità degli osteoblasti, riducendo così la frequenza delle fratture, consiste nell’armatura delle ossa lunghe. Attualmente sono già in uso strumentazioni intelligenti (cioè allungabili) per la correzione della deformità ossea, intra-midollari per le ossa lunghe e sul rachide per la scoliosi (6,8,20-22). Sin dagli anni ‘70 e ’80 c‘è stata indicazione al trattamento chirurgico delle scoliosi severe, mediante artrodesi strumentata posteriore, in particolare nei casi in cui la scoliosi mostra una rapida evoluzione in età precoce e il trattamento conservativo non ha successo. L'alta incidenza di complicanze, dovute alle difficoltà di ancoraggio dello strumentario per la ridotta resistenza meccanica del tessuto osseo, ha reso prudente negli anni l’approccio chirurgico alle deformità della colonna. Solo recentemente, grazie all’evoluzione degli strumentari (fig 6) e delle tecniche chirurgiche e anestesiologiche, gli interventi per scoliosi in questa patologia sono diventati meno rischiosi e con soddisfacente correzione intra-operatoria (6,23-26).
Ipofosfatasia
Genetica: mutazione del gene codificante per la fosfatasi alcalina. Ne esistono tre tipi, il III diagnosticabile in utero e con prognosi peggiore.
Incidenza: molto rara.
Clinica: micromelia, ipoplasia toracica, ipomineralizzazione, idrope ed edema nei casi diagnosticati precocemente.
SRPS (Short Rib Polydactyl Syndrome)
Eziologia: sconosciuta.
Incidenza: molto rara.
Clinica: se ne riconoscono due forme, entrambe con le stesse caratteristiche ecografiche, spesso associate a cardiopatia congenita. Micromelia, ipoplasia toracica, esadattilia.
Esito: letale nel 100% dei casi.
Displasia camptomelica
Genetica: mutazione del gene codificante la proteina SRY-box 9, presente nel testicolo e nello scheletro.
Incidenza: rara (0.2/10mila nati).
Clinica: rizomelia, tibia e femore incurvati, ipoplasia delle scapole, micrognazia, sex reversal nei maschi.
Esito: quasi sempre letale.
DISPLASIE SCHELETRICHE NON LETALI
Displasia distrofica
Genetica: mutazione del gene DTDST.
Incidenza: molto rara.
Clinica: deformità posturali, micrognazia, contratture degli arti (pollice da “autostoppista”).
Esito: letale nel 25% dei casi, grave handicap fisico per le contratture e la cifoscoliosi in coloro che sopravvivono.
Acondroplasia.
Genetica: mutazione del gene codificante per Fibroblast Growth Factor Receptor. Se ne distinguono due sottotipi: omozigote ed eterozigote.
Incidenza: relativamente frequente (1/10mila nati).
Clinica: rizomelia, tendenza alla macrocrania, naso insellato, mano a tridente.
Esito: sopravvivenza e performance mentale nella norma, problematiche ortopediche e polmonari a lungo termine.
Distrofia toracica asfissiante
Eziologia: sconosciuta.
Incidenza: rara.
Clinica: ipoplasia toracica, anomalie renali, rizomelia moderata.
Esito: letale nel 60% dei casi per ipoplasia polmonare.
Distrofia condro-ectodermica
Genetica: mutazione locus 4p16 che porta ad anomalia del tessuto di accrescimento cartilagineo tra epifisi e diafisi e a displasia dei tessuti di origine ectodermica.
Incidenza: molto rara (1/60mila nati).
Clinica: acro-mesomelia, ipoplasia toracica di media entità, esadattilia post-assiale, soprattutto delle mani, cardiopatia congenita (difetto inter-atriale).
Esito: letale nel 50% dei casi.
Distrofia metatropica
Eziologia: mutazione nel gene TRPV4, che codifica per un canale cationico permeabile al Ca2+ presente in diversi tessuti. Le mutazioni possono provocare un aumento del calcio nei condrociti e, di conseguenza, un'alterazione dell'ossificazione endo-condrale ed i segni clinici.
Incidenza: non nota (ad oggi sono stati descritti circa 81 casi) (1).
Clinica: displasia spondilo-epimetafisaria, caratterizzata da torace lungo e arti corti nel periodo neonatale, successiva cifo-scoliosi grave e progressiva, che esita in inversione delle proporzioni nell'infanzia (torace corto e arti lunghi) e statura finale bassa nell'età adulta. Lo spettro fenotipico è variabile e si associa a forme gravi con esito letale "in utero", o immediatamente dopo la nascita, e a forme che presentano solo lievi alterazioni scheletriche.
Diagnosi: si basa sui segni clinici e radiologici. Questi variano a seconda dell'età e comprendono diafisi corte con metafisi larghe, significativa plati-spondilia, calcificazione precoce della cartilagine ioide e cricoide, bacino a forma di alabarda, grave ipoplasia della parte anteriore delle prime vertebre cervicali, con anomalie e aspetto squadrato delle ossa del calcagno. I test molecolari possono identificare le mutazioni diTRPV4, confermando la diagnosi.
Esito: la prognosi varia a seconda della gravità della malattia. L'attesa di vita non è di solito compromessa, a meno che non vi siano complicazioni respiratorie.
CONCLUSIONI
Il percorso diagnostico delle displasie scheletriche è fondamentalmente clinico-radiologico: la valutazione accurata dei segni clinici e radiologici può consentire almeno l’inquadramento in uno dei 42 gruppi o famiglie e indirizzare a eventuali indagini genetiche mirate.
Negli ultimi vent’anni sono stati compiuti notevoli progressi nella comprensione dei disturbi correlati a mutazioni del recettore FGF-R3 e dei meccanismi patogenetici alla base di tali disturbi, in modo da delineare strategie terapeutiche efficaci nel trattamento dei difetti di crescita ossea associati a questo tipo di mutazioni. Sebbene ci sia stato un certo successo nello sviluppo di nuove terapie, il miglioramento ulteriore del trattamento di bambini e adulti affetti rappresenta tuttora una sfida affascinante. Ci sono diverse nuove strategie terapeutiche che potranno essere prese in considerazione in futuro. Inoltre, sarà importante studiare il potenziale sinergismo di due o più inibitori farmacologici dell’FGF-R3 o della via di segnale a valle, che potrebbero aumentare l’efficacia del trattamento. L’eterogeneità clinica e allelica di diverse condizioni, in relazione a mutazioni in domain diversi di uno stesso gene e soprattutto in malattie estremamente rare, può complicare l’iter diagnostico. L’attuale livello di conoscenze ha reso possibile la diagnosi con diversi criteri clinici, radiologici e molecolari della maggior parte delle 461 displasie scheletriche attualmente note. In questi casi, per giungere alla diagnosi è indispensabile ricorrere a un’indagine molecolare con sequenziamento completo dell'esoma, che consente di definire la diagnosi nella quasi totalità dei casi (92%). Nello stesso tempo sono molto migliorate le condizioni di vita dei pazienti grazie ai progressi in campo terapeutico, sia chirurgico, per prevenire le deformità, sia farmacologico con il ricorso a farmaci nuovi o già in uso per altre indicazioni, sia con la terapia enzimatica sostitutiva (ad es. in alcune mucopolisaccaridosi), ma soprattutto con terapie specifiche (trial già conclusi o in itinere nei casi di acondroplasia).
BIBLIOGRAFIA
- Scarano G, Falco M, Scarano F, et al. Bassa statura e sindromi rare: le displasie scheletriche. Archivio MR 2020 - 2.
- Tsang KY, Tsang SW, Chan D, Cheah KSE. The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Def Res (Part C) 2014, 102: 52–73.
- Spranger J, Maroteaux P. The lethal osteochondrodysplasias. Adv Hum Genet 1990, 19: 1-103, 331-2.
- Stanescu V, Stanescu R, Maroteaux P. Pathogenic mechanisms in osteochondrodysplasias. J Bone Joint Surg Am 1984, 66: 817-36.
- Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal of genetic skeletal disorders: 2019 revision. Am J Med Genet 2019, 179A: 2393-419.
- Bonafè L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet Part A 2015, 167: 2869-92.
- Hoover-Fong J, Scott CI, Jones MC; Committee on Genetics. Health supervision for people with achondroplasia. Pediatrics 2020, 145: e20201010.
- Schiedel F, Rödl R. Lower limb lengthening in patients with disproportionate short stature with achondroplasia: a systematic review of the last 20 years. Disabil Rehabil 2012, 34: 982-7.
- Donaldson J, Aftab S, Bradish C. Achondroplasia and limb lengthening: results in a UK cohort and review of the literature. J Orthop 2015, 12: 31-4.
- Kim SJ, Pierce W, Sabharwal S. The etiology of short stature affects the clinical outcome of lower limb lengthening using external fixation. A systematic review of 18 trials involving 547 patients. Acta Orthop 2014, 85: 181-6.
- Semler O, Rehberg M, Mehdiani N, et al. Current and emerging therapeutic options for the management of rare skeletal diseases. Paediatr Drugs 2019, 21: 95-106.
- Wang J, Zhou J, Cheng CM, et al. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol 2004, 180: 247–55.
- Gorbenko O, Ovcharenko G, Klymenko T, et al. Generation of monoclonal antibody targeting fibroblast growth factor receptor 3. Hybridoma (Larchmt) 2009, 28: 295-300.
- Gust KM, McConkey DJ, Awrey S, et al. Fibroblast growth factor receptor 3 is a rational therapeutic target in bladder cancer. Mol Cancer Ther 2013, 12: 1245-54.
- Liu Z, Lavine KJ, Hung IH, Ornitz DM. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol 2007, 302: 80-91.
- Garcia S, Dirat B, Tognacci T, et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci Transl Med 2013, 5: 203ra124.
- Breinholt VM, Rasmussen CE, Mygind PH, et al. TransCon CNP, a sustained-release C-type natriuretic peptide prodrug, a potentially safe and efficacious new therapeutic modality for the treatment of comorbidities associated with fibroblast growth factor receptor 3-related skeletal dysplasias. J Pharmacol Exp Ther 2019, 370: 459-71.
- Argente J, Martos Moreno GA. Skeletal dysplasias: new medical treatments. An Pediatr (Barc) 2016, 85: 1-3.
- Greggi T. Esperienza e stabilizzazione vertebrale nell'osteogenesi imperfetta. L'osteogenesi imperfetta: il percorso diagnostico, terapeutico e assistenziale dal bambino all’adulto. Congresso Nazionale Associazione Italiana Osteogenesi Imperfetta Onlus - Milano 2-5 maggio 2013.
- Wallace MJ, Kruse RW, Shah SA. The spine in patients with osteogenesis imperfecta. J Am Acad Orthop Surg 2017, 25: 100-9.
- Keuning MC, Leeuwerke SJG, van Dijk PR, et al. Prevalence of scoliosis and impaired pulmonary function in patients with type III osteogenesis imperfecta. Eur Spine J 2022, 31: 2295-300.
- Burnei G, Vlad C, Georgescu I, et al. Osteogenesis imperfecta: diagnosis and treatment. J Am Acad Orthop Surg 2008, 16: 356-66.
- Paley D. PRECICE intramedullary limb lengthening system. Expert Rev Med Devices 2015, 12: 231-49.
- Piantoni L, Noel MA, Francheri Wilson IA, et al. Surgical treatment with pedicle screws of scoliosis associated with osteogenesis imperfecta in children. Spine Deform 2017, 5: 360-5.
- Karlin LI, McClung A, Johnston CE, et al; Children’s Spine Study Group. The growth-friendly surgical treatment of scoliosis in children with osteogenesis imperfecta using distraction-based instrumentation. Spine Deform 2021, 9: 263-74.
- Gardner A, Sahota J, Dong H, et al. The use of magnetically controlled growing rods in paediatric osteogenesis imperfecta. J Surg Case Rep 2018, 2018: rjy043.
Scheda inibitori aromatasi
Fedra Mori
UOC di Endocrinologia, Azienda Ospedaliera Sant’Andrea, Roma
Meccanismo d’azione
L’aromatasi è un enzima che appartiene alla famiglia del citocromo P450 ed è espresso ubiquitariamente nelle cellule umane (es. cellule della granulosa, cellule di Leydig, tessuto adiposo, cervello, fibroblasti, sincizio-trofoblasto) (1,2).
La sua espressione nelle cellule della granulosa è fondamentale per il processo di sintesi dell’estradiolo, poichè permette la conversione dell’androstenedione in estrone. In menopausa l’espressione principale dell’aromatasi si trova invece a livello del tessuto adiposo.
Gli inibitori dell’aromatasi quindi riducono la sintesi degli estrogeni in tutti i tessuti e trovano applicazione nel trattamento di tutte quelle condizioni che sono estrogeno-dipendenti (3).
Negli anni si è passati da una prima generazione di inibitori dell’aromatasi (aminoglutetimide) gravati da un importante effetto collaterale rappresentato dall’insufficienza surrenalica, a una seconda generazione costituita da farmaci che devono essere somministrati im, a quelli più recenti, di cosiddetta terza generazione (letrozolo, anastrozolo, exemestano), che sono somministrati per via orale e appaiono selettivi, potenti e reversibili (3).
Indicazioni
Cancro della mammella. Questa rappresenta al momento l’unica indicazione approvata per l’uso di questi farmaci. La terapia adiuvante con inibitori dell’aromatasi sembra ridurre l’incidenza di cancro controlaterale, di invasività e carcinoma duttale in situ nelle donne in menopausa e positività del recettore per estrogeni (4-7). Questa rappresenta al momento l’unica indicazione approvata per questa classe di farmaci che possono essere prescritti ormai (det. AIFA 30.10.12; GU n. 267 del 15-11-2012) senza piano terapeutico e senza alcuna nota. Gli inibitori dell’aromatasi vengono utilizzati anche nel trattamento del carcinoma mammario del maschio (8).
Endometriosi. Il trattamento con analoghi di terza generazione si è dimostrato efficace nel ridurre il dolore pelvico. Possono essere utilizzati inoltre in associazione con altri farmaci quali gli analoghi del GnRH, con una migliore soppressione dei livelli estrogenici (9).
Induzione dell’ovulazione. Sebbene al momento non siano raccomandati routinariamente, vi sono in letteratura numerose evidenze che identificano gli inibitori dell’aromatasi come farmaci utili nell’induzione dell’ovulazione in donne affette da PCO o infertilità inspiegata, con effetti comparabili al clomifene citrato in termini di safety e rischio di gravidanze multiple (3,10).
In pediatria gli inibitori dell’aromatasi hanno trovato diverse applicazioni off-label (11):
- Sindrome di Mc Cune Albright: gli studi attualmente a disposizione hanno dimostrato solo una parziale e transitoria efficacia del letrozolo nel controllare la progressione puberale delle bambine affette da questa sindrome (12,13);
- Testotossicosi: migliori risultati in termini di riduzione della velocità di crescità e maturazione ossea sembrano derivare dall’uso contemporaneo di anti-androgeni (bicalutamide, ciproterone) associati a letrozolo o anastrozolo (14,15);
- Bassa statura idiopatica: poichè gli estrogeni hanno un ruolo importante sia nell’induzione dello spurt puberale che nella maturazione e chiusura delle epifisi, gli inibitori dell’aromatasi sono stati utilizzati, da soli o in associazione con altri farmaci, nel trattamento di bambini con bassa statura idiopatica o ritardo puberale costituzionale. Sebbene alcuni studi dimostrino un vantaggio in termini di statura finale nei bambini trattati rispetto ai controlli, ci sono anche evidenze di un incremento delle deformità vertebrali nei trattati e mancano studi dei possibili effetti collaterali a lungo termine (11,16,17).
Contro-indicazioni
Pazienti in pre-menopausa, poiché a causa del ridotto feed-back degli estrogeni, inducono un aumento della secrezione delle gonadotropine e in alcuni esperimenti su animali determinano un aumento delle dimensioni e del peso delle ovaie. Le pazienti con carcinomi mammari che risultano privi di recettori per gli estrogeni sono usualmente non responsive ai trattamenti ormonali.
Preparazioni, via di somministrazione, posologia
- Anastrozolo: cp 1 mg (Adiunastrol, Anastrozolo Alter, Anastrozolo Aurobindo, Anastrozolo DOC generici, Anastrozolo EG, Anastrozolo Mylan, Anastrozolo Pensa, Anastrozolo Sandoz, Anastrozolo Sun, Anastrozolo TEVA, Anastrozolo Zentiva, Antabrest, Arimidex, Eristrol, Extroplex, Griset, Raoloz, Renazole)
- Letrozolo: cp 2.5 mg (Brestoral, Calantha, Femara, Gosuran, Letrix, Letrozolo Almus, Letrozolo Alter, Letrozolo Aurobindo, Letrozolo DOC generici, Letrozolo EG, Letrozolo Mylan Generics, Letrozolo Pensa, Letrozolo Sandoz, Letrozolo Sun, Letrozolo Technigen, Letrozolo TEVA, Letrozolo Zentiva, Raziolet, Zolobrest, Zoltron)
- Exemestane: cp 25 mg (Aromasin, Axelta, Exegen, Exemestane DOC Generici, Exemestane EG, Exemestane Mylan Generics, Exemestane Sandoz, Exemestane TEVA, Exemestane Zentiva, Mestane, Mexabrest, Naxestan, Nibestan).
Effetti collaterali
Effetti collaterali a medio e lungo temine: raramente vampate, secchezza vaginale, modesta cefalea, dolore e tumefazione articolare, compresa la sindrome del tunnel carpale. I disturbi articolari sembrano responsabili di circa il 10% dei casi di interruzione precoce della terapia (18).
L’uso prolungato può indurre, in entrambi i sessi, osteopenia e osteoporosi, con incremento del rischio di frattura (19,20). L'effetto negativo osseo cessa immediatamente alla sospensione della terapia (21). L’uso di bisfosfonati è in grado di prevenire completamente la perdita minerale ossea indotta dagli inibitori (22).
Possibile peggioramento del profilo lipidico, sebbene non sia stata dimostrato un incremento del rischio cardiovascolare in confronto al tamoxifene (23).
Limitazioni prescrittive
Senza piano terapeutico e senza nota per il carcinoma mammario.
Bibliografia
- Simpson ER, et al. Aromatase gene expression in adipose tissue: relationship to breast cancer. Int J Fertil Menopausal Stud 1994, 39 suppl 2: 75–83.
- Grodin JM, et al. Source of estrogen production in postmenopausal women. J Clin Endocrinol Metab 1973, 36: 207–14.
- Pavone ME, Bulun SE. The use of aromatase inhibitors for ovulation induction and superovulation. J Clin Endocrinol Metab 2013, 98: 1838–44.
- Baum M, et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet 2002, 359: 2131–9.
- Goss PE, et al. A randomized trial of letrozole in postmenopausal women after five years of tamoxifen therapy for early-stage breast cancer. N Engl J Med 2003, 349: 1793–802.
- Goss PE, et al. Exemestane for breast-cancer prevention in postmenopausal women. N Engl J Med 2011, 364: 2381–91.
- Advani P, et al. Current strategies for the prevention of breast cancer. Breast Cancer 2014, 6: 59–71.
- Bradley KL, et al. Contemporary systemic therapy for male breast cancer. Clin Breast Cancer 2014, 14: 31-9.
- Pavone ME, Bulun SE. Aromatase inhibitor for the treatment of endometriosis: a review. Fertil Steril 2012, 98: 1370–9.
- Eckmann KR, Kockler DR. Aromatase inhibitors for ovulation and pregnancy in polycystic ovary syndrome. Ann Pharmacother 2009, 43: 1338-4.
- Wit JM, et al. Aromatase inhibitors in pediatrics. Nat Rev Endocrinol 2012, 8: 135–47.
- Feuillan P, et al. Letrozole treatment of precocious puberty in girls with the McCune–Albright syndrome: a pilot study. J Clin Endocrinol Metab 2007, 92: 2100–6.
- Mieszczak J, et al. The aromatase inhibitor anastrozole is ineffective in the treatment of precocious puberty in girls with McCune–Albright Syndrome. J Clin Endocrinol Metab 2008, 93: 2751–4.
- Lenz AM, et al. Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics 2010, 126: e728–33.
- Eyssette-Guerreau S, et al. Effectiveness of anastrozole and cyproterone acetate in two brothers with familial male precocious puberty. J Pediatr Endocrinol Metab 2008, 21: 995–1002.
- Diaz-Thomas A, et al. Use of aromatase inhibitors in children and adolescents: what’s new? Curr Opin Pediatr 2010, 22: 501–7.
- Ranke MB. Treatment of children and adolescents with idiopathic short stature. Nat Rev Endocrinol 2013, 9: 325-34.
- Adjuvant aromatase inhibitors in early breast cancer – toxicity and adherence. Important observations in clinical practice. Breast Cancer Res Treatm 2007, 106 (S1): S111.
- Buzdar AU. The ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial: an update. Clin Breast Cancer 2004, 5 suppl 1: S6–12.
- Jakesz R, et al. Switching of postmenopausal women with endocrine-responsive early breast cancer to anastrozole after 2 years' adjuvant tamoxifen: combined results of ABCSG trial 8 and ARNO 95 trial. Lancet 2005, 366: 455–62.
- Eastell R, et al. Effect of anastrozole on bone mineral density: 5-year results from the anastrozole, tamoxifen, alone or in combination trial 18233230. J Clin Oncol 2008, 26: 1051–8.
- Gnant M, et al. Adjuvant endocrine therapy plus zoledronic acid in premenopausal women with early-stage breast cancer: 5-year follow-up of the ABCSG-12 bone-mineral density substudy. Lancet Oncol 2008, 9: 840–9.
- Geisler J, et al. Changes in bone and lipid metabolism in postmenopausal women with early breast cancer after terminating 2-year treatment with exemestane: a randomised, placebo-controlled study. Eur J Cancer 2006, 42: 2968–75.
Ginecomastia nell'adolescenza
Antonio Radicioni, Antonella Valente, Matteo Spaziani
SMID04 Diagnostica ormonale, Seminologia e Banca del seme
Azienda Policlinico Umberto I – Sapienza Università di Roma
(aggiornato al 21 gennaio 2020)
Introduzione
Per ginecomastia, dal greco gynec (donna) e mastos (petto), si intende l’aumento di volume della ghiandola mammaria nel maschio, determinato dalla proliferazione benigna di elementi duttali del parenchima ghiandolare (1). Da questa condizione clinica va distinta l’adipomastia (detta anche lipomastia o pseudo-ginecomastia), caratterizzata da un’eccessiva presenza di tessuto adiposo in regione mammaria, frequentemente, ma non esclusivamente, presente nei soggetti obesi. La ginecomastia può essere bilaterale o monolaterale (2).
Nell’adolescente frequentemente determina un importante senso d’ansia, distress per la propria immagine corporea e riduzione della qualità di vita: un nostro paziente di 14 anni con un promettente futuro nel nuoto nazionale, dopo diversi anni e ottimi risultati agonistici, aveva deciso di abbandonare il nuoto per le difficoltà di mostrarsi in costume.
Prima della pubertà è possibile osservare una forma neonatale abbastanza rara: il passaggio trans-placentare di estrogeni materni e la conversione di DHEA e DHEAS ad estrone (E1) ed estradiolo (E2) da parte del tessuto placentare determinano uno sbilanciamento del rapporto E2/T nel feto. È usualmente una forma benigna, ad evoluzione spontanea dopo qualche settimana dalla nascita. Se la ginecomastia persiste per più di un anno, devono essere indagate altre possibili cause sottostanti.
I meccanismi alla base della ginecomastia includono una diminuzione della produzione di androgeni, l’aumento della produzione di estrogeni o la maggior disponibilità di precursori degli estrogeni per la conversione periferica. Altre possibili cause sono il blocco del recettore degli androgeni e, in pazienti con alterata produzione di testosterone (T), la ridotta concentrazione di T libero (fTe) dovuta all’aumento della SHBG. La stimolazione da parte degli estrogeni determina iperplasia duttale, allungamento e diramazione dei dotti ghiandolari, proliferazione dei fibroblasti peri-duttali e incremento della vascolarizzazione (1). Tutte le condizioni che aumentano la concentrazione di SHBG possono determinare riduzione di fTe.
Molti studi effettuati su maschi in età puberale con ginecomastia non hanno mostrato alterazioni della concentrazione sierica di T, E2, E1 e gonadotropine rispetto a maschi di pari età senza ginecomastia. Tuttavia, in alcuni casi si osserva un transitorio rapido incremento di E2 all’inizio della pubertà nei ragazzi che sviluppano ginecomastia. Questi mostrano talvolta ampie fluttuazioni dei livelli di E2, con aumento della concentrazione di E2 totale nelle 24h, che riflette un aumento della conversione degli androgeni surrenalici in estrogeni. L’incremento della concentrazione di E2 fino a livelli dell’adulto si verifica sempre prima dell’aumento della concentrazione di T: è proprio in questa fase che può generarsi lo squilibrio del rapporto E2/T (3). Leptina e IGF-1 sono aumentati sia nei ragazzi con ginecomastia, sia in quelli senza. Tuttavia, l’evidenza che la ginecomastia puberale si sviluppa in associazione a un rapido raggiungimento del picco di concentrazione di queste due molecole, evidenzia che effettivamente sia la leptina che l’IGF-1 possono essere implicate nello sviluppo della ginecomastia puberale (4).
L’incidenza della ginecomastia, durante l’età evolutiva, nei lavori della letteratura varia notevolmente, fra 4 e 69%, in base al diverso modo di valutare l’incremento ghiandolare e alle diverse età considerate dagli autori. Risulta estremamente rara prima dei 10 anni, aumenta con il progredire della maturazione puberale, con un picco fra 13 e 14 anni nella fase medio avanzata della pubertà (stadio puberale di Tanner G3-G4).
Diagnosi
Usualmente il giovane paziente o i genitori riferiscono di aver notato il rigonfiamento in regione mammaria in una fase estremamente critica dello sviluppo puberale, pertanto giungono all’osservazione con molta ansia e preoccupazione.
Nella ginecomastia dell’adolescente l’iter diagnostico prevede il dosaggio di gonadotropine, T, E2, SHBG, PRL, TSH e fT4. Nel caso di una sostanziale normalità di questi dosaggi di primo livello, certamente non si può escludere la possibilità che i ragazzi con ginecomastia potrebbero presentare un più basso rapporto di delta4-androstenedione/E1 e DHEAS/E2, dovuto a un’aumentata aromatizzazione negli adolescenti obesi, quindi con una temporanea alterazione della conversione degli androgeni surrenalici ad estrogeni. Questa condizione potrebbe non essere rilevabile a livello sistemico, se si realizzasse temporaneamente solo a livello locale, con alterato rapporto tra gli effetti stimolatori degli estrogeni e quelli inibitori degli androgeni sul tessuto mammario.
L’obesità o il sovrappeso possono favorire la comparsa della ginecomastia, perché il tessuto adiposo della regione mammaria contiene l’enzima aromatasi, che converte T e delta-4 rispettivamente in E2 ed E1, ma la ginecomastia si può osservare anche in ragazzi normopeso e in rari casi anche sottopeso.
L’ecografia bilaterale della regione mammaria è uno strumento molto utile per valutare il volume della componente ghiandolare e del tessuto adiposo (tab. 1).
| Tabella 1 Classificazione ecografica (5) |
||
| Tipo | Descrizione | Risposta alla terapia medica |
| Florida | Iperplasia e proliferazione dei dotti, con aumento ed edema dello stroma | Buona |
| Intermedia | Caratteristiche intermedie | |
| Fibrosa | Fibrosi diffusa dello stroma | Scarsa/assente |
Ginecomastia patologica o secondaria
In epoca peri-puberale si può sviluppare una ginecomastia secondaria a patologie diverse, come l’ipogonadismo primario e secondario, in particolare la sindrome di Klinefelter, patologie epatiche croniche, tumori testicolari, ipertiroidismo e ipotiroidismo, alterazioni della differenziazione sessuale, e anche all’uso di alcuni farmaci e sostanze che possono aumentare la concentrazione di PRL.
In tutte le forme di ipogonadismo si assiste a un incremento del rapporto E2/T, che favorisce lo sviluppo della ginecomastia, la quale può anche rappresentare il sintomo di esordio. L’ipogonadismo primario è caratterizzato da una ridotta produzione di T, aumentati livelli di LH, con aumentata aromatizzazione del T ad E2. Per contro, nell’ipogonadismo secondario i ridotti livelli di T sono dovuti alla riduzione della secrezione di LH, in presenza di produzione di precursori degli estrogeni da parte del surrene (fig 1). Quindi, in entrambe le condizioni si verifica l’incremento del rapporto E2/T.
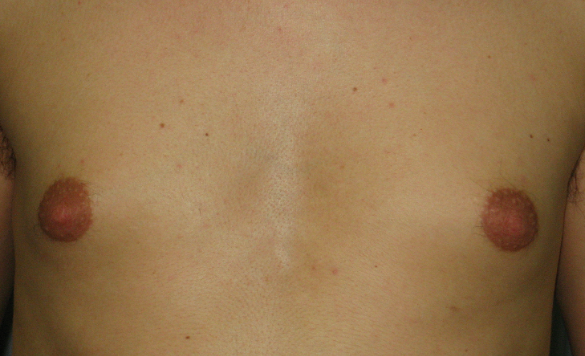
Figura 1. Ginecomastia e adipomastia in paziente di 16.4 anni con ipogonadismo ipogonadotropo
La sindrome di Klinefelter (KS) rappresenta la più comune anomalia cromosomica associata ad ipogonadismo. La prevalenza della ginecomastia in questi pazienti è compresa tra 50 e 70% nelle diverse casistiche e merita particolare attenzione del clinico, perché può permettere di sospettare l’alterazione cromosomica e anche per l’aumentato rischio di sviluppare un carcinoma mammario in questa patologia (6) (fig 2).
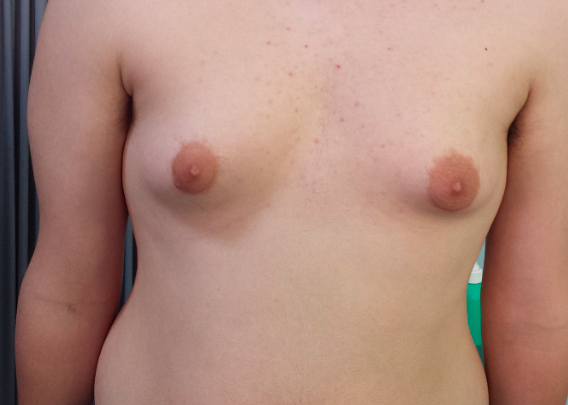
Figura 2. Ginecomastia in paziente con s. di Klinefelter di 15.2 anni
L’iperprolattinemia può essere implicata nella patogenesi della ginecomastia come causa secondaria di ipogonadismo. Tuttavia, nel tessuto mammario maschile sono stati trovati recettori per PRL (7), che possono essere co-espressi e cross-regolati con i recettori del GH e del progesterone (PGR). L’attivazione del PGR è spesso associata a una riduzione del recettore per gli androgeni (AR): ciò ostacola l’inibizione della crescita del tessuto mammario mediata dal T, che si osserva in condizioni di normale omeostasi ormonale. Di conseguenza, oltre che inducendo ipogonadismo, l’iperprolattinemia può favorire lo sviluppo di ginecomastia attraverso una via completamente differente: elevati livelli di PRL possono stimolare la crescita del tessuto mammario come risultato di eccessiva attivazione del PGR e ridotta disponibilità dell’AR.
La ginecomastia può essere anche il primo segno di un tumore testicolare. Tumori testicolari a cellule germinali inducono ginecomastia mediante l’incremento dei livelli di βhCG da parte della neoplasia (coriocarcinoma o foci di cellule trofoblastiche all’interno di un seminoma) (8). Gli alti livelli di βhCG che ne derivano, all’interno delle cellule di Leydig alterano l’attività enzimatica della 17-idrossilasi e incrementano l’attività dell’aromatasi testicolare, con incremento della conversione di T ad E2 (9). Infine, la ginecomastia si sviluppa anche nel 15% dei pazienti trattati con successo per neoplasia testicolare (orchiectomia, chemio/radioterapia): l’impatto che la chemioterapia e la radioterapia possono avere sul testicolo contro-laterale può determinare ipogonadismo secondario, che spesso si risolve spontaneamente nell’arco di un anno. Per contro, la ginecomastia può comparire nel 20-30% delle neoplasie testicolari non a cellule germinali. Il tumore a cellule di Leydig determina il diretto incremento della secrezione di estrogeni e/o l’incremento dell’attività dell’aromatasi testicolare. Anche i tumori a cellule di Sertoli sono associati allo sviluppo di ginecomastia e femminilizzazione, tramite l’aumento dell’attività dell’aromatasi testicolare.
La ginecomastia può comparire in adolescenti affetti da ipertiroidismo, per l’aumento di E2, SHBG e T. L’aumento dei livelli di LH è inoltre responsabile dell’aumentata aromatizzazione di T in E1-E2. Anche l’ipotiroidismo può favorire lo sviluppo di ginecomastia per l’aumentata secrezione di TRH e conseguentemente di PRL. Sia in ipertiroidismo che in ipotiroidismo, la ginecomastia può essere risolta ristabilendo lo stato di eutiroidismo.
Studi non recenti hanno permesso di dimostrare in adolescenti che sia l’aumento dei livelli di leptina che i polimorfismi nel suo recettore possono essere coinvolti nella patogenesi della ginecomastia: il metabolismo accelerato degli estrogeni e l'aumento della espressione dell’aromatasi sono stati identificati come fattori che inducono ginecomastia. Anche l'asse GH-IGF potrebbe avere un ruolo nella patogenesi di questa patologia (10).
C'è una solida evidenza che molti farmaci sono in grado di indurre ginecomastia (11). In epoca puberale si debbono ricordare rhGH, anti-psicotici, corticosteroidi, oppioidi e dopanti, che debbono essere attentamente ricercati nell’anamnesi di questi ragazzi (12,13).
Esame obiettivo locale
Il giovane paziente deve essere visitato in clino- e orto-statismo. Pur tenendo conto della classificazione di Rohrich (tab 2), riteniamo più semplice e riproducibile la misura del diametro medio della lesione mammaria di consistenza aumentata e granulosa indicativa del nucleo ghiandolare. Deve essere previsto un esame generale, con particolare attenzione a quello genitale, per valutare lo sviluppo puberale ed eventuali sospetti di ipogonadismo.
| Tabella 2 Classificazione clinica (14) |
||
| Grado | Ipertrofia | Ptosi |
| I | Minima (< 250 g) | No |
| II | Moderata (250-500 g) | No |
| III | Severa (> 500 g) | I grado |
| IV | II-III grado | |
Terapia
In linea generale la decisione di trattare la mammella dipende dalla scelta del paziente e dall’impatto che la patologia ha sulla sua qualità di vita.
Nei casi di ginecomastia fisiologica, dovuta spesso a uno squilibrio ormonale transitorio, non è necessario nessun trattamento: nell’adolescente la malattia può regredire spontaneamente nel giro di 12-18 mesi.
È importante cercare di identificare le forme secondarie e iatrogene. Nel caso in cui la ginecomastia non si risolva spontaneamente ed evolva verso quadri non più regredibili, si rende necessario il trattamento medico e/o chirurgico, che viene quindi riservato soltanto a una ristretta percentuale di pazienti adolescenti.
Ulteriori indicazioni a trattare sono le elevate dimensioni delle mammelle (> 4 cm) (fig 3) e/o la presenza di dolore mammario.
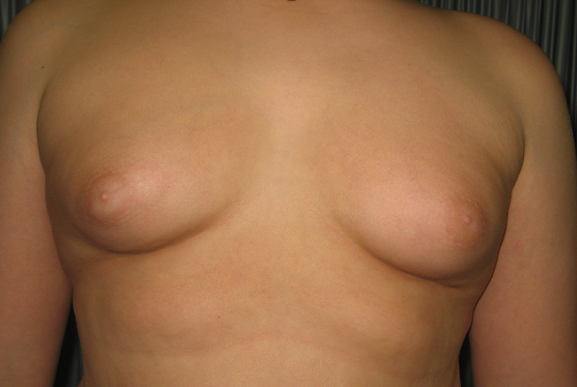
Figura 3. Paziente di 16.1 anni, obeso e ipotiroideo con ginecomastia
Una volta identificate e trattate le cause della ginecomastia, sono necessari alcuni mesi prima di assistere alla riduzione del volume mammario. Nei pazienti con ginecomastia dolorosa che non possono essere sottoposti ad intervento chirurgico e che non sono candidati ad altre terapie, può essere utile il trattamento con anti-estrogeni, come il tamoxifene (20 mg/die), in grado di ridurre il dolore e le dimensioni mammarie in più della metà dei pazienti. In studi su un limitato numero di pazienti il tamoxifene o il raloxifene, antagonisti del recettore degli estrogeni, hanno ridotto le dimensioni della ghiandola, anche se è poco frequente una regressione completa della ginecomastia. Poiché la base delle prove è attualmente bassa, la prescrizione di tamoxifene resta off-label e ciò deve essere attentamente spiegato ai pazienti (15). Anche gli inibitori dell'aromatasi possono essere efficaci, specie nelle fasi iniziali della malattia. In uno studio randomizzato su uomini con ginecomastia confermata, l’anastrozolo non si è però dimostrato più efficace del placebo nel ridurre le dimensioni mammarie (16).
Vanno inviati alla chirurgia tutti quegli adolescenti che mostrino/riferiscano importanti conseguenze sulla qualità di vita e accettazione della propria immagine corporea, in cui le terapie mediche non hanno permesso di ottenere risultati, anche di tipo estetico, accettabili per il giovane paziente. L’intervento di escissione del parenchima ghiandolare e del tessuto adiposo della regione mammaria, se eseguito da chirurgo con specifiche competenze, può permettere di ottenere risultati, sul piano estetico, molto interessanti e utili per i nostri adolescenti.
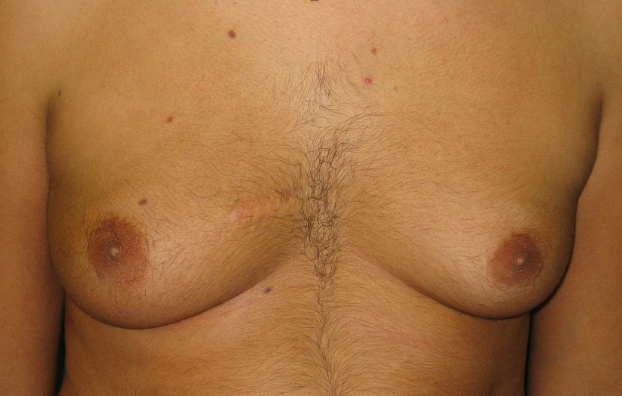
Figura 4. Ginecomastia in paziente di 17.3 anni, stadio puberale G5, con ipogonadismo primitivo ma valori di T nei limiti della norma ed aumento del rapporto E2/T
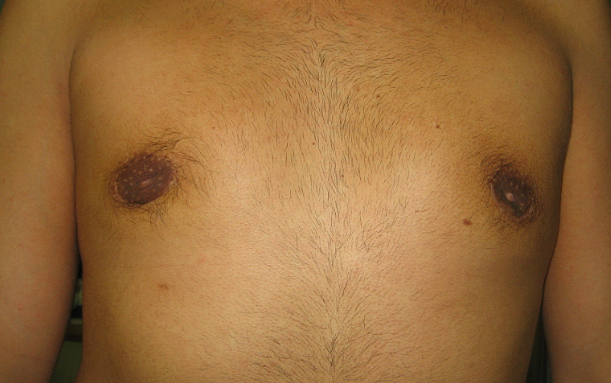
Figura 5. Esiti di intervento per ginecomastia bilaterale: evidenti le cicatrici peri-areolari
Bibliografia
- Cuhaci N, Polat SB, Evranos B, et al. Gynecomastia: clinical evaluation and management. Indian J Endocrinol Metab 2014, 18: 150–8.
- Johnson RE, Murad MH. Gynecomastia: pathophysiology, evaluation, and management. Mayo Clin Proc 2009, 84: 1010–5.
- Moore DC, Schlaepfer LV, Paunier L, Sizonenko PC. Hormonal changes during puberty: V. Transient pubertal gynecomastia: abnormal androgen-estrogen ratios. J Clin Endocrinol Metab 1984, 58: 492-9.
- Limony Y, Friger M, Hochberg Z. Pubertal gynecomastia coincides with peak height velocity. J Clin Res Pediatr Endocrinol 2013, 5: 142-4.
- Bannayan GA, Hajdu SI. Gynecomastia: clinicopathologic study of 351 cases. Am J Clin Pathol 1972, 57: 431-7
- Radicioni AF, Ferlin A, Balercia G, et al. Consensus statement on diagnosis and clinical management of Klinefelter syndrome J Endocrinol Invest 2010, 33: 839-50.
- Ferreira M, Mesquita M, Quaresma M, Andre S. Prolactin receptor expression in gynaecomastia and male breast carcinoma. Histopathology 2008, 53: 56–61.
- Kayemba-Kays S, Fromont-Hankard G, Lettelier G, et al. Leydig cell tumour revealed by bilateral gynecomastia in a 15-year-old adolescent: a patient report. J Pediatr Endocrinol Metab 2010, 23: 1195–9.
- Huang Y, Song J, Xu M, Zan Q. Primary Leydig cell tumour of epididymis: a rare case report with review of literature. Andrologia 2013, 45: 430–3.
- Dundar B, Dundar N, Erci T, et al. Leptin levels in boys with pubertal gynecomastia. J Pediatr Endocrinol Metab 2005, 18: 929–34.
- Deepinder F, Braunstein GD. Drug-induced gynecomastia: an evidence-based review. Expert Opin Drug Saf 2012, 11: 779–95.
- Nieschlag E, Vorona E. Mechanisms in endocrinology: medical consequences of doping with anabolic androgenic steroids: effects on reproductive functions. Eur J Endocrinol 2015, 173: R47–58.
- Eckman A, Dobs A. Drug-induced gynecomastia. Expert Opin Drug Saf 2008, 7: 691–702.
- Rohrich RJ, Ha Ry, Kenkel JM, Adams WP Jr. Classification and management of gynecomastia: defining the role of ultrasound-assisted liposuction. Plast Reconstr Surg 2003, 111: 909-23.
- Staiman VR, Lowe FC. Tamoxifen for flutamide/finasteride-induced gynecomastia. Urology 1997, 50: 929-33.
- Plourde PV, Reiter EO, Jou HC, et al. Safety and efficacy of anastrozole for the treatment of pubertal gynecomastia: a randomized, double-blind, placebo-controlled trial. J Clin Endocrinol Metab 2004, 89: 4428-33.