Approccio terapeutico alla pubertà ritardata

Rossella Gaudino, con la collaborazione di Sarah Dal Ben
Dipartimento di Scienze della Vita e Riproduzione, UO Complessa Pediatria, Policlinico Verona, AOUI di Verona
Endocrinologia e Auxologia Pediatrica e dell'Adolescenza
In tutti i casi di ritardo puberale l'obiettivo del trattamento è lo sviluppo dei caratteri sessuali secondari, l'accelerazione della velocità di crescita, il miglioramento della statura definitiva e, se possibile, il raggiungimento della capacità riproduttiva. La terapia inoltre agisce anche sul contesto psico-sociale del bambino, migliorandone la stima e i rapporti con gli altri (1).
La terapia del ritardo puberale varia in base alla diversa eziologia.
Nelle forme secondarie a patologie croniche e/o deficit nutrizionali si tratta la causa sottostante.
Nel caso in cui, invece, la causa sia un deficit gonadico, la terapia è ormonale sostitutiva con ormoni sessuali, i cui dosaggi (tabella 1 e 2) vengono aumentati progressivamente ogni tre anni per mimare il normale percorso di accrescimento.
Nelle forme di ipogonadismo ipogonadotropo la terapia ormonale sostitutiva non induce nè la crescita testicolare nè l'ovulazione, perciò l'induzione dello sviluppo in entrambi i sessi necessita di gonadotropine esogene o GnRH (2-4).
Per quanto concerne il CDGP, il trattamento può prevedere semplicemente l'osservazione del paziente o, in casi selezionati, una terapia ormonale con testosterone (nel maschio) o estrogeni (nella femmina) (tabella 1 e 2). Molto importante sia nella vigile attesa che nella terapia è offrire un supporto psicologico: i pazienti, infatti, spesso sono preoccupati per l'altezza che raggiungeranno e l'aspetto fisico. Recenti studi nei maschi hanno evidenziato come la terapia ormonale sostitutiva nel CDGP abbia risvolti positivi non solo sulla crescita ma anche sull'aspetto psicologico (5-7).
Per quanto riguarda l'uso del GH, esso non è raccomandato nei pazienti con CDGP in quanto non migliora l'altezza finale (1).
Un'altra opzione terapeutica nei maschi con CDGP e bassa altezza potrebbe essere l'utilizzo di inibitori dell'aromatasi, che permetterebbero di prolungare la crescita lineare e di raggiungere un'altezza finale maggiore (8,9). Il loro utilizzo, in ogni caso, non è ancora stato approvato, visti i possibili effetti collaterali a carico dell'osso (10).
| Tabella 1 Trattamento ormonale sostitutivo nel maschio (1) |
|||||
| Principio attivo | CDGP | Ipogonadismo | Effetti collaterali | ||
| enantato, cipropionato e propionato (im) | Non raccomandato prima dei 14 anni Dose iniziale 50-100 mg ogni 4 settimane per 3-6 mesi Il trattamento va ripetuto con aumenti delle dose di 15-50 mg |
Dopo i 12 anni si può iniziare con 50 mg ogni 4 settimane, con incrementi di 50 mg ogni 6-12 mesi Al raggiungimento del dosaggio mensile di 100-150 mg va somministrato ogni 2 settimane La dose dell'adulto è 200 mg ogni 2 settimane |
Eritrocitosi, aumento di peso, iperplasia prostatica | ||
| undecanoato | Dose dell'adulto 1000 mg ogni 10-14 giorni | ||||
| Inibitori dell'aromatasi | letrozolo po | 2.5 mg/die | Non raccomandato | Diminuzione HDL, eritrocitosi, deformità vertebrali | |
| anastrozolo po | 1 mg/die | Non raccomandato | |||
| Pompa sottucutanea: rilascio pulsatile GnRH | Non raccomandato | Inizialmente: 5-25 ng/kg/pulse ogni 90-120 min; mantenimento: 25-600 ng/kg/pulse | |||
| hCG e rhFSH (sottocutaneo) | Non raccomandato | hCG: 500-3000 UI inizialmente due volte alla settimana, poi ogni due giorni rhFSH: 75-225 UI due/tre volte alla settimana. |
hCG: possibile apoptosi delle cellule germinali. Nell'HH è necessario l'uso di FSH per indurre la crescita testicolare e la spermatogenesi | ||
| Tabella 2 Trattamento ormonale sostitutivo nella donna (1) |
|||
| Principio attivo | CDGP | Ipogonadismo |
Effetti collaterali |
| Etinil-estradiolo | Dose iniziale 2 μg/die Aumentare a 5 μg/die dopo 6-12 mesi |
Dose iniziale 2 μg/die Aumentare ogni 6-12 mesi a 5-10-20 μg/die |
Tossicità epatica, aumento del rischio di trombo-embolismo, ipertensione arteriosa |
| 17β-estradiolo (somministrazione orale) | Dose iniziale 5 μg/kg/die per via orale Aumentare a 10 μg/kg/die dopo 6-12 mesi |
Dose iniziale 5 μg/kg/die per via orale Aumentare ogni 6-12 mesi a 10-15-20 μg/kg/die Dose dell'adulto 1-2 mg/die |
|
| 17β-estradiolo (via trans-dermica) | Dose iniziale 3.1-6.2 μg/24h Aumentare di 3.1-6.2 μg/24h ogni 6 mesi |
Dose iniziale 3.1-6.2 μg/24h Aumentare di 3.1-6.2 μg/24h ogni 6 mesi Dose dell'adulto 50-100 μg/24h |
|
| Estrogeni equini coniugati (CEE) (somministrazione orale) | Dose iniziale 0.1625 mg/die per 6-12 mesi, poi aumentare a 0.325 mg/die | Dose iniziale 0.1625 mg/die per 6-12 mesi, poi aumentare ogni 6-12 mesi a 0.325-0.45-0.625 mg/die | Riportato aumento del rischio cardiovascolare |
| Necessario solo se il trattamento dura più di 12 mesi | 5-10 mg/die di medrossiprogesterone acetato (MPA) negli ultimi 7 giorni del ciclo mestruale In alternativa 100-200 μg/die di progesterone |
Il progesterone aggiunto agli estrogeni induce il ciclo endometriale | |
| GnRH pulsatile | Non raccomandato | Usato per la fertilità | |
BIBLIOGRAFIA
- Palmert MR, Dunkel L. Clinical practice. Delayed puberty. N Engl J Med 2012, 366: 443-53.
- Pitteloud N, Hayes FJ, Dwyer A, et al. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2002, 87: 4128-36.
- Pitteloud N, Hayes FJ, Boepple PA, et al. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2002, 87: 152-60.
- Warne DW, Decosterd G, Okada H, et al. A combined analysis of data to identify predictive factors for spermatogenesis in men with hypogonadotropic hypogonadism treated with recombinant human follicle-stimulating hormone and human chorionic gonadotropin. Fertil Steril 2009, 92: 594-604.
- Soliman AT, Khadir MM, Asfour M. Testosterone treatment in adolescent boys with constitutional delay of growth and development. Metabolism 1995, 44: 1013-5.
- Richman RA, Kirsch LR. Testosterone treatment in adolescent boys with constitutional delay in growth and development. N Engl J Med 1988, 319: 1563-7.
- Rosenfeld RG, Northcraft GB, Hintz RL. A prospective, randomized study of testosterone treatment of constitutional delay of growth and development in male adolescents. Pediatrics 1982, 69: 681-7.
- Hero M, Norjavaara E, Dunkel L. Inhibition of estrogen biosynthesis with a potent aromatase inhibitor increases predicted adult height in boys with idiopathic short stature: a randomized controlled trial. J Clin Endocrinol Metab 2005, 90: 6396-402.
- Wickman S, Sipila I, Ankarberg-Lindgren C, et al. A specific aromatase inhibitor and potential increase in adult height in boys with delayed puberty: a randomised controlled trial. Lancet 2001, 357: 1743-8.
- Hero M, Toiviainen-Salo S, Wickman S, et al. Vertebral morphology in aromatase inhibitor-treated males with idiopathic short stature or constitutional delay of puberty. J Bone Miner Res 2010, 25: 1536-43.
Overview sulle resistenze ormonali
Marco Bonomi
Divisione Endocrinologia e Metabolismo, Istituto Auxologico Italiano, Milano
Negli ultimi anni sono stati fatti importanti progressi nell’ambito delle resistenze ormonali. L’avanzamento delle nuove tecnologie applicate alla genetica e all’epigenetica ha permesso di migliorare in modo rilevante le conoscenze riguardanti la patogenesi delle malattie pediatriche dovute a resistenza ormonale e, di conseguenza, le nostre capacità diagnostiche in quest’ambito. L’attenta caratterizzazione del fenotipo di questi pazienti con resistenza ormonale, unitamente ai risultati della ricerca traslazionale, ha migliorato la nostra capacità di cura dei pazienti affetti.
Le patologie da resistenza ormonale sono condizioni causate da una ridotta o assente risposta d’organo a ormoni biologicamente attivi e possono essere dovute a un difetto del recettore ormonale (per esempio glucocorticoidi, androgeni, estrogeni, ormone paratiroideo, ormone antidiuretico, insulina), sia esso di membrana o nucleare, o a un difetto post-recettoriale. Indipendentemente dalla diversa resistenza ormonale e dal meccanismo recettoriale implicato, tutti questi quadri presentano un’azione ormonale deficitaria, nonostante siano presenti concentrazioni circolanti di ormone normali o superiori alla norma. La prima descrizione di resistenza all’azione ormonale risale alla metà del secolo scorso da parte di Albright, che descrisse le caratteristiche cliniche dei pazienti con pseudoipoparatiroidismo (1). Da allora sono stati descritti molti altri quadri di resistenza ormonale, così come elencato nella tabella.
| Principali forme di resistenza all’azione ormonale | ||
| Recettore | Sindrome | Fenotipo clinico |
| TRH | Resistenza al TRH | Ipotiroidismo centrale |
| TSH | Resistenza al TSH | Ipotiroidismo congenito |
| TR | Resistenza agli ormoni tiroidei | Resistenza all’azione degli ormoni tiroidei |
| GnRH | Resistenza al GnRH | Ipogonadismo Ipogonadotropo Isolato |
| LH | Ridotta sensibilità a LH | Pseudoermafroditismo maschile |
| FSH | Resistenza a FSH | Disgenesia ovarica ipergonadotropa |
| AR | Resistenza agli androgeni | Femminilizzazione testicolare Pseudoermafroditismo maschile |
| ESR1 | Resistenza agli estrogeni | Resistenza agli estrogeni |
| AMH | Resistenza ad AMH | Sindrome da persistenza del dotto di Muller |
| ACTH | Resistenza all’ACTH | Deficit familiare di glucocorticoidi |
| GR | Resistenza ai glucocorticoidi | Ipertensione con eccesso di mineralcorticoidi |
| Aldosterone | Resistenza all’aldosterone | Pseudoiperaldosteronismi |
| ADH | Resistenza all’ADH | Diabete insipido nefrogenico |
| GHRH | Resistenza al GHRH | Nanismo, deficit di GH |
| GH | Resistenza al GH (s. di Laron) | Nanismo |
| GNAS | Pseudoipoparatiroidismo | Variabili |
| VDR | Resistenza alla Vitamina D | Rachitismo ereditario Vit D-resistente |
| CaSR | Ridotta sensibilità al calcio | Ipercalcemia familiare ipocalciurica Iperparatiroidismo primitivo neonatale |
| Insulina | Resistenza insulinica | Diabete mellito tipo 2 |
Sebbene le sindromi da resistenza all’azione ormonale abbiano una base ereditaria, gli individui affetti, anche all’interno della medesima famiglia, possono presentare una variabilità fenotipica. D’altro canto, nella stessa sindrome si può assistere a un’eterogeneità genetica e molecolare, come pure a livello della cellula bersaglio. Nella trattazione delle sezioni a seguire saranno forniti elementi di valutazione clinica e genetica delle diverse resistenze ormonali, illustrando come difetti molecolari legati alla produzione ormonale, piuttosto che alle loro vie di segnale intra-cellulari o ai meccanismi di risposta, possano essere alla base di malattie negli esseri umani.
Le sindromi da resistenza ormonale possono colpire i diversi assi ipotalamo-ipofisari, a partire da quello che regola la funzione tiroidea. In questo ambito sono note la resistenza al TRH e al TSH, che si presentano con specifici quadri da ipotiroidismo. La resistenza al TRH può essere legata non solo ad alterazioni del recettore del TRH ma, come recentemente dimostrato, anche a forme legate al cromosoma X a seguito di alterazioni a carico del gene IGSF1 (2). La resistenza al TSH può presentare quadri clinici di entità variabile, che vanno da un ipotiroidismo congenito importante nelle forme complete a una semplice ipertireotropinemia non autoimmune con sintomi sfumati di ipotiroidismo nelle sue forme parziali con difetto genetico in eterozigosi semplice (3). Un’altra resistenza ormonale nell’asse ipotalamo-ipofisi-tiroide è rappresentata dalla resistenza all’azione degli ormoni tiroidei (4). Tale resistenza era storicamente legata a varianti del recettore ß degli ormoni tiroidei. Tuttavia è del tutto recente la descrizione di uno specifico quadro clinico di resistenza all’azione degli ormoni tiroidei legato a varianti del recettore alfa (4).
L’asse ipotalamo-ipofisi surrene presenta specifici quadri di resistenza ormonale, quali la resistenza all’ACTH e ai glucocorticoidi (6,7). Entrambe queste forme di resistenza ormonale possono portare a quadri clinici di estrema gravità, che necessitano una pronta diagnosi e terapia.
Le resistenze ormonali legate al corretto funzionamento dell’asse ipotalamo-ipofisi gonadi sono rappresentate dalle forme centrali di resistenza al GnRH (8) e dalla forme da resistenza alle gonadotropine (9) o agli steroidi sessuali (10,11). I quadri clinici che ne derivano sono particolarmente importanti per lo sviluppo sessuale e la fertilità degli individui affetti.
L’asse GH-IGF-I presenta a sua volta forme di resistenze ormonali che interferiscono a livello recettoriale o post-recettoriale, alterando il regolare processo di accrescimento corporeo (12,13).
Abbiamo poi resistenze ormonali anche a carico del metabolismo fosfo-calcico, che possono interessare sia il PTH, con meccanismi genetici ed epigenetici (1) che la vitamina D (14), determinando una forma ereditaria di rachitismo.
Infine la forma di resistenza ormonale più comune è quella rappresentata dalla resistenza insulinica, che si associa a quadri di alterato metabolismo come nell’obesità e/o nel diabete mellito di tipo 2 (15).
La trattazione di questa specifica sezione fornirà informazioni utili al clinico per la diagnosi di queste forme recettoriali che, in alcuni casi, sono relativamente rare e quindi poco conosciute. Il corretto riconoscimento dei diversi quadri clinici sarà la base successiva per una terapia mirata al quadro di resistenza diagnosticato.
Bibliografia
- Mantovani G. Pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab 2011, 96: 3020-30.
- Persani L. Central hypothyroidism: pathogenic, diagnostic, and therapeutic challenges. J Clin Endocrinol Metab 2012, 97: 3068-78.
- Beck-Peccoz P, Persani L, Calebiro D, et al. Syndromes of hormone resistance in the hypothalamic-pituitary-thyroid axis. Best Pract Res Clin Endocrinol Metab 2006, 20: 529-46.
- Refetoff S, Bassett JH, Beck-Peccoz P, et al. Classification and proposed nomenclature for inherited defects of thyroid hormone action, cell transport, and metabolism. J Clin Endocrinol Metab 2014, 99: 768-70.
- Meimaridou E, Hughes CR, Kowalczyk J, et al. ACTH resistance: genes and mechanisms. Endocr Dev 2013, 24: 57-66.
- Meimaridou E, Hughes CR, Kowalczyk J, et al. Familial glucocorticoid deficiency: new genes and mechanisms. Mol Cell Endocrinol 2013, 371: 195-200.
- van Rossum EF, van den Akker EL. Glucocorticoid resistance. Endocr Dev 2011, 20: 127-36.
- Bonomi M, Libri DV, Guizzardi F, et al; Idiopathic Central Hypogonadism Study Group of the Italian Societies of Endocrinology and Pediatric Endocrinology and Diabetes. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl 2012, 14: 49-56.
- Latronico AC, Arnhold IJ. Gonadotropin resistance. Endocr Dev 2013, 24: 25-32.
- Smith EP, Specker B, Bachrach BE, et al. Impact on bone of an estrogen receptor-alpha gene loss of function mutation. J Clin Endocrinol Metab 2008, 93: 3088-96.
- Quaynor SD, Stradtman EW Jr, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med 2013, 369: 164-71.
- Alatzoglou KS, Dattani MT. Genetic causes and treatment of isolated growth hormone deficiency-an update. Nat Rev Endocrinol 2010, 6: 562-76.
- Wit JM, Oostdijk W, Losekoot M. Spectrum of insulin-like growth factor deficiency. Endocr Dev 2012, 23: 30-41.
- Feldman D, J Malloy P. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. Bonekey Rep 2014, 3: 510.
- Paneni F, Costantino S, Cosentino F. Insulin resistance, diabetes, and cardiovascular risk. Curr Atheroscler Rep 2014, 16: 419.
Pseudoipoparatiroidismo
Giovanna Mantovani
Dipartimento di Scienze Cliniche e di Comunità - Università degli Studi di Milano, UO Endocrinologia e Malattie Metaboliche, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano
Introduzione, fisiopatologia e genetica
Lo Pseudoipoparatiroidismo (PHP) e l’Osteodistrofia Ereditaria di Albright (Albright hereditary osteodystrophy - AHO) (OMIM 103580) sono patologie correlate, rare, potenzialmente disabilitanti con una componente genetica ormai accertata. La loro prevalenza è stimata intorno a 0.79/100.000 (Orphanet Report Series, 2011).
Lo PHP rappresenta storicamente la prima sindrome da resistenza ormonale, descritta nel 1942 da Fuller Albright (1): in realtà il termine comprende un gruppo eterogeneo di rare malattie metaboliche, caratterizzate da resistenza periferica all’azione del PTH con conseguente ipocalcemia e iperfosforemia in presenza di elevati livelli di PTH (tab. 1) (2,3).
Già nella sua prima descrizione Albright riportava in questi pazienti la presenza di una funzione renale conservata, associata però a ridotta risposta calcemica e fosfaturica dopo iniezione di estratto bovino di paratiroide, soprattutto se confrontata con quella osservata nei pazienti con ipoparatiroidismo primitivo. Studi successivi hanno poi dimostrato la coesistenza di iperplasia delle paratiroidi con elevazione del PTH serico, confermando pertanto l’esistenza di quella che oggi definiamo “resistenza ormonale” (1-3).
L’identificazione del recettore del PTH e della sua via di trasduzione del segnale mediata dal legame con la proteina G etero-trimerica stimolatoria (Gs), con conseguente produzione di AMP ciclico (cAMP), ha poi permesso di comprendere la fisiopatologia della malattia e di identificare anche una ridotta secrezione di cAMP nel siero e nelle urine dei soggetti affetti dopo iniezione di PTH bovino. Questo ha anche consentito di differenziare il PHP di tipo I, che presenta la tipica ridotta risposta sia fosfaturica che in termini di cAMP, dal PHP tipo II, in cui la risposta fosfaturica è ridotta mentre rimane conservata quella di cAMP, a indicare un difetto intra-cellulare distale rispetto alla generazione di cAMP. Ad oggi, sono relativamente poche le descrizioni di casi certi di PHP-II e il difetto molecolare rimane ignoto. È stato anche ipotizzato che questa non sia in realtà una patologia genetica, ma rappresenti un difetto acquisito secondario a severa ipovitaminosi D, dal momento che in molti pazienti la sua supplementazione insieme alla somministrazione di calcio è in grado di ripristinare una risposta fosfaturica fisiologica al PTH esogeno (2).
La tabella 1 schematizza l’inquadramento del PHP tipo I.
| Tabella 1 Classificazione dello pseudoipoparatiroidismo tipo I |
||||||
| Sottotipo | AHO | Resistenze ormonali | Trasmissione | Risposta all’infusione di PTH | Attività Gs | Difetto di GNAS |
| Ia | Sì | PTH/TSH/GHRH/ Gonadotropine | AD | Assente | Ridotta | Genetico (trasmissione materna) |
| Pseudo | Sì | No | AD | Normale | Ridotta | Genetico (trasmissione paterna) |
| Ib | No | PTH/TSH | AD/Sporadica | Assente | Normale/ Ridotta | Epigenetico |
| Ic | Sì | PTH/TSH/ GHRH/ Gonadotropine | AD se associato a mutazioni di GNAS | Assente | Normale | Pochi casi riportati con difetto genetico |
I due sottotipi principali di PHP, PHP Ia e Ib, sono causati da alterazioni genetiche all'interno o a monte del locus GNAS, che dà origine anche al gene che codifica per la Gs-alfa (subunità alfa della proteina Gs). Circa il 70% dei pazienti con PHP-Ia, che presentano AHO associata a resistenza all’azione di diversi ormoni che attivano le vie accoppiate alla Gs, quali PTH, TSH, gonadotropine e GHRH, presenta mutazioni in eterozigosi di derivazione materna negli esoni 1-13 di GNAS. Le stesse mutazioni ereditate dal padre causano invece lo pseudo-pseudoipoparatiroidismo (pseudo-PHP)(OMIM 612463). Questo termine viene usato per indicare la condizione di pazienti con AHO. L’osteodistrofia ereditaria di Albright comprende un quadro clinico eterogeneo, caratterizzato da brachidattilia, viso rotondeggiante, bassa statura, obesità, ritardo mentale e ossificazioni sottocutanee (tab. 2).

Le fotografie mostrano una bambina con AHO: in alto si può vedere la bassa statura e la facies tipica; in basso le mani con le dita corte e le calcificazioni sottocutanee

I pazienti PHP-Ib non mostrano AHO e la resistenza ormonale è limitata a PTH e, in misura minore, TSH, in assenza dei classici segni di AHO; il meccanismo patogenetico alla base della patologia è la perdita di elementi per il controllo dell’imprinting a lungo raggio del locus GNAS (perdita sporadica o su base genetica), con conseguente diminuzione della trascrizione della Gs alfa nei tubuli renali prossimali e resistenza al PTH.
Il quadro di PHP-Ic è clinicamente indistinguibile da quello Ia, ma, con pochissime eccezioni, sono assenti le tipiche mutazioni a carico del gene codificante la Gs-alfa, così come è normale l’attività della proteina Gs misurata negli eritrociti o nelle piastrine di questi pazienti.
Nella pratica clinica, sia l’osteodistrofia che la resistenza ormonale sono spesso difficili da diagnosticare, in quanto le caratteristiche cliniche non sono evidenti alla nascita e possono poi diventare molto eterogenee, con anomalie endocrine che si verificano in età diverse e con gravità variabile. Durante lo scorso decennio, nuovi dati hanno reso ancor più difficile la distinzione tra le diverse malattie associate a GNAS. Infatti, in un sottogruppo di pazienti con PHP e gradi variabili di AHO, sono stati identificati difetti epigenetici di GNAS simili a quelli associati allo PHP-Ib, suggerendo l’esistenza di un overlap molecolare tra PHP-Ia e PHP-Ib. Inoltre, sono state identificate mutazioni nei geni PRKAR1A e PDE4D, codificanti proteine cruciali per il segnale intra-cellulare mediato dal cAMP, in pazienti con acrodisostosi, una patologia ossea con caratteristiche simili all’AHO che può anche essere associata a pluri-resistenza ormonale. Inoltre, mutazioni di GNAS di derivazione paterna causano l’eteroplasia ossea progressiva (POH), in cui le ossificazioni ectopiche si estendono progressivamente nel muscolo scheletrico e nei tessuti connettivi profondi.
Complessivamente, la sovrapposizione molecolare e clinica tra questi disordini evidenzia la necessità di nuovi modelli di classificazione e modifica profondamente la nostra conoscenza dei meccanismi con cui i difetti della via di trasduzione del segnale mediata dal cAMP causano PHP/AHO/POH. Inoltre, nonostante l’avanzamento delle tecniche diagnostiche, circa il 25% dei pazienti rimane ancora oggi senza una diagnosi molecolare.
Gestione clinica e terapia
Nella pratica clinica l’AHO è talvolta di difficile inquadramento, in quanto alcuni segni non sono evidenti alla nascita e possono essere altamente eterogenei più avanti nel tempo. Nel caso della presenza di PHP, le analisi di laboratorio ormonali sono spesso dirimenti, ma possono anch’esse essere confondenti, dal momento che anche le alterazioni endocrine possono divenire evidenti in epoche della vita diverse ed essere molto variabili in termini di gravità.
Devono essere eseguiti annualmente un attento esame obiettivo e, quando necessario, una specifica valutazione psicologica, allo scopo di scoprire e seguire la presenza o l’evoluzione di specifiche caratteristiche di AHO (in particolare le ossificazioni ectopiche e il ritardo mentale). Lo screening iniziale dovrebbe includere la valutazione radiologica della brachidattilia.
In generale, i pazienti affetti da PHP-I devono essere monitorati annualmente, con il dosaggio di PTH, calcemia totale e ionizzata, calciuria, fosforemia, 25-OH-vitamina D, TSH. Inoltre, nei bambini deve essere posta un’attenzione particolare al monitoraggio dell’altezza, del peso, della velocità di crescita e dello sviluppo puberale.
Recenti evidenze suggeriscono che, indipendentemente dalla curva di crescita, i bambini debbano essere valutati con test di provocazione per il deficit di GH, allo scopo di iniziare il trattamento il prima possibile (4-7). Devono essere determinati peso e BMI allo scopo di iniziare, quando necessario, un programma dietetico o di esercizio fisico.
I dati disponibili relativi alla mineralizzazione ossea, sembrano finora indicare un quadro di normalità, a indicare la “resistenza” anche dell’osso alla cronica esposizione a livelli estremamente elevati di PTH.
| Tabella 2 Criteri diagnostici |
|
| Esami di laboratorio | |
| Criteri maggiori | Ipocalcemia, iperfosforemia e livelli elevati di PTH in assenza di ipovitaminosi D (quadro di resistenza al PTH) |
| Addizionali | Livelli elevati di TSH, in assenza di evidenza sierica ed ecografica di autoimmunità tiroidea (resistenza al TSH) Livelli elevati di LH e FSH, associati a estradiolo/testosterone ridotti (resistenza alle gonadotropine) Ridotta risposta del GH ai test di stimolo (resistenza al GHRH) |
| Segni clinici | |
| Maggiori (associati all’ipocalcemia acuta o cronica) | Ipereccitabilità nervosa con parestesie, crampi, tetania, iper-reflessia, convulsioni, crisi tetaniche Cataratta Calcificazioni dei nuclei della base |
| Addizionali |
Amenorrea/oligomenorrea e/o infertilità
|
La terapia a lungo termine dell’ipocalcemia nei pazienti affetti da PHP è simile a quella delle altre forme di ipoparatiroidismo. Il trattamento con metaboliti attivi della vitamina D, preferibilmente calcitriolo con o senza supplemento di calcio per os, deve mirare a mantenere una condizione di normocalcemia. La terapia dovrebbe essere sufficientemente aggressiva da mantenere la calcemia nel mid-range di normalità, senza dovere necessariamente normalizzare o avvicinare alla normalità i valori di PTH sierici. Raramente (generalmente in età infantile) può essere necessaria l’aggiunta di chelanti del fosforo per ridurre l’iperfosforemia. Nell’aggiustare il dosaggio di calcio e vitamina D, bisogna considerare che questi pazienti hanno un basso rischio di aumentare l’escrezione di calcio urinario sotto terapia con calcitriolo, pertanto il trattamento può generalmente essere più aggressivo rispetto ai pazienti con ipoparatiroidismo.
Nei pazienti con PHP-I devono essere sistematicamente valutate ed eventualmente trattate le endocrinopatie associate, in particolare l’ipotiroidismo e l’ipogonadismo. La levo-tiroxina e gli ormoni sessuali dovrebbero essere somministrati seguendo gli stessi criteri, dosi e follow-up delle altre forme di ipotiroidismo e ipogonadismo. Non ci sono ad oggi studi sistematici sulla fertilità in questi pazienti, né tantomeno sull’esito di fecondazione medicalmente assistita.
Dalla prima dimostrazione della presenza di deficit di GH nei pazienti con PHP-Ia (4-5), poi confermato da studi più recenti (6), non esistono dati conclusivi sulla necessità di una terapia sostitutiva con GH. Infatti, la rilevanza effettiva del deficit di GH sull’altezza finale in questa patologia rimane incerta, essendo la bassa statura di questi pazienti più probabilmente il risultato di una somma di fattori multipli, tra i quali gioca un ruolo preponderante la fusione prematura delle cartilagini di accrescimento, con conseguente assenza dello spurt puberale. Tuttavia, uno studio pilota condotto su 8 bambini con PHP-Ia trattati con GH in età prepubere, indica un significativo incremento nella velocità di crescita, quantomeno prima della pubertà, suggerendo che il trattamento con rhGH debba essere iniziato il prima possibile a causa della finestra temporale relativamente breve per una terapia potenzialmente efficace (7). In particolare, l’osservazione su un’unica paziente che l’altezza finale può essere aumentata ritardando l’esposizione dell’osso agli estrogeni tramite l’utilizzo di analoghi del GnRH, sottolinea la necessità di studi più ampi e accurati per stimolare gli endocrinologi a diagnosticare e trattare correttamente e precocemente questi pazienti al fine di ottenere i massimi benefici possibili.
Infine, non esistono trattamenti specifici per le varie manifestazioni di AHO e il trattamento chirurgico delle ossificazioni ectopiche va limitato ai casi in cui queste risultino particolarmente estese o fastidiose e potenzialmente limitanti per la vicinanza alle articolazioni.
Bibliografia
- Albright F, Burnett CH, Smith CH, Parson W. Pseudohypoparathyroidism: an example of “Seabright-Bantam syndrome”. Endocrinology 1942, 30: 922-32.
- Mantovani G. Clinical review: Pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab 2011, 96: 3020-30.
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev 2001, 22: 675-705.
- Mantovani G, Maghnie M, Weber G, et al. Growth hormone-releasing hormone resistance in pseudohypoparathyroidism type Ia: new evidence for imprinting of the Gs alpha gene. J Clin Endocrinol Metab 2003, 88: 4070-4.
- Germain-Lee EL, Groman J, Crane JL, et al. Growth hormone deficiency in pseudohypoparathyroidism type 1a: another manifestation of multihormone resistance. J Clin Endocrinol Metab 2003, 88: 4059-69.
- de Sanctis L, Bellone J, Salerno M, et al. GH secretion in a cohort of children with pseudohypoparathyroidism type Ia. J Endocrinol Invest 2007, 30: 97-103.
- Mantovani G, Ferrante E, Giavoli C, et al. Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type Ia: first study on the effect on growth. J Clin Endocrinol Metab 2010, 95: 5011-7.
Resistenza ai glucocorticoidi
Alessandra Vottero & Roberta Minari
Università degli Studi di Parma
Definizione, epidemiologia e fisiopatologia
La resistenza ai glucocorticoidi (GC) è una sindrome rara ed estremamente eterogenea, caratterizzata da una parziale o generalizzata insensibilità dei tessuti all’azione dei glucocorticoidi. Può essere transitoria o permanente, parziale o completa e compensata o non compensata. Recentemente questa condizione è stata rinominata come “sindrome di Chrousos”, dal nome del medico che per primo l’ha descritta.
Nell’uomo i glucocorticoidi regolano numerosi processi biologici critici, quali la crescita, la riproduzione, il metabolismo, le reazioni immunologiche e infiammatorie, così come le funzioni cardiovascolari e quelle che riguardano il sistema nervoso centrale. Inoltre, giocano un ruolo importante nel mantenimento dell’omeostasi basale e correlata allo stress.
A livello cellulare le funzioni dei GC si esplicano attraverso il legame con il loro recettore (GCR), una proteina di 94 kDa che appartiene alla superfamiglia dei recettori nucleari e di cui si conoscono due isoforme: l’isoforma alfa è quella classica che media le funzioni dei GC, mentre la ß esercita un effetto dominante negativo sull’attività trascrizionale di alfa.
Nell’ambito della resistenza ai GC, sono state descritte diverse anomalie genetiche dei GCR, rappresentate principalmente da mutazioni inattivanti. In presenza della perdita funzionale del recettore, si determina un’attivazione compensatoria dell’asse, responsabile di un’aumentata secrezione di CRH e arginina-vasopressina (AVP) nel sistema portale ipofisario, e conseguentemente di ACTH nella circolazione sistemica. L’eccessiva secrezione di ACTH determina iperplasia surrenalica e conseguentemente aumentata secrezione di mineral-corticoidi (principalmente DOC e corticosterone) e androgeni (DHEA, DHEA-S e androstenedione).

Fisiopatologia (modif da 2): a sinistra la situazione fisiologica, a destra le modificazioni indotte dalla sindrome
Clinica
Sulla base della severità della resistenza ai GC, le manifestazioni cliniche possono variare notevolmente, passando da condizioni asintomatiche a segni e sintomi da eccesso di mineralcorticoidi, come ipertensione e/o alcalosi ipokaliemica, e ad iperandrogenismo.
Nel bambino, la secrezione precoce ed eccessiva degli androgeni surrenalici può manifestarsi come genitali ambigui alla nascita in un neonato con cariotipo femminile, pubarca prematuro isolato e pubertà precoce gonadotropina-indipendente. Nelle donne determina acne, irsutismo, calvizie, irregolarità mestruali, mentre nell’uomo sono stati osservati oligospermia e infertilità, probabilmente dovuti a disturbi della regolazione dell’FSH causati dall’eccesso di androgeni surrenalici.
In alcuni pazienti può manifestarsi una profonda ansia, causata probabilmente dall’aumento della secrezione ipotalamica di CRH e AVP, che può anche causare lo sviluppo di un adenoma ACTH-secernente. Infine, i livelli elevati di ACTH possono essere responsabili della crescita di tessuto surrenalico testicolare. Tuttavia, un’elevata percentuale di pazienti con resistenza ai GC è asintomatica e presenta solo alterazioni biochimiche.
Le manifestazioni cliniche da deficit di glucocorticoidi sono molto rare e sono state riportate solamente in un bambino piccolo con ipoglicemia generalizzata e convulsioni tonico-cloniche durante un episodio febbrile, in un neonato con severa ipoglicemia, eccessivo affaticamento ad alimentarsi, aumentata suscettibilità alle infezioni e concomitante deficit di GH, e in alcuni pazienti adulti con fatica cronica.
| Manifestazioni cliniche in relazione al meccanismo fisio-patologico | ||
| Funzione gluco-corticoide apparentemente normale | Forma asintomatica Sindrome da affaticamento cronico |
|
| Eccesso di mineral-corticoidi | Ipertensione Alcalosi ipokaliemica |
|
| Eccesso di androgeni | Età pediatrica | Genitali ambigui alla nascita Pubarca prematuro isolato Pubertà precoce |
| Femmine | Acne Irsutismo Calvizie Irregolarità mestruali con cicli oligo-anovulatori Infertilità |
|
| Maschi | Acne Irsutismo Oligospermia Tessuto surrenalico testicolare Infertilità |
|
| Ipersecrezione di CRH/ACTH | Ansia Depressione |
|
Diagnosi
Per sospettare una resistenza ai glucocorticoidi è indispensabile una dettagliata anamnesi familiare e personale del paziente, con particolare attenzione ad alterazioni nell’attività dell’asse ipotalamo-ipofisi-surrene.
Il ritmo secretorio circadiano di cortisolo è conservato, così come la risposta allo stress, mentre si osserva un’insufficiente soppressione del cortisolo al test di soppressione con desametasone (sia con la dose singola di 1 mg a mezzanotte, dose da aggiustare per superficie corporea nel bambino, che con le dosi multiple).
Il cortisolo libero urinario è aumentato e le concentrazioni plasmatiche di ACTH risultano normali o aumentate.
| Valutazioni diagnostiche |
| Assenza di caratteristiche Cushingoidi Livelli plasmatici di ACTH normali o elevati Livelli plasmatici di cortisolo elevati Aumentata escrezione di cortisolo libero urinario nelle 24h Normale ritmo circadiano e stress-indotto di cortisolo e ACTH Resistenza dell’asse ipotalamo-ipofisi-surrene alla soppressione con desametasone Studi molecolari: mutazioni/delezioni del gene GR |
Le ghiandole surrenaliche possono presentarsi debolmente aumentate di volume.
La diagnosi si basa sulla presenza di ipercortisolismo in assenza di caratteristiche Cushingoidi, in quanto i test standard utilizzati per l’ipercortisolismo non discriminano tra resistenza ai GC e sindrome di Cushing.
Inoltre, a causa della secrezione eccessiva di androgeni surrenalici, la densità minerale ossea è generalmente ai limiti alti della norma o elevata in questi pazienti, a differenza di quello che si osserva nella sindrome di Cushing, dove, invece, un segno importante è rappresentato dall’osteoporosi.
Trattamento
Il trattamento di questi pazienti consiste nella somministrazione di dosi personalizzate di desametasone (1-3 mg/die per os), potente gluco-corticoide sintetico con minima attività mineralcorticoide. Il desametasone, a queste dosi farmaceutiche, sopprime l’ACTH e di conseguenza anche cortisolo endogeno, DOC, corticosterone e androgeni surrenalici, correggendo lo stato di eccesso di mineral-corticoidi e androgeni caratteristico di questi pazienti. In generale, negli adulti si può iniziare con una dose di 1 mg di desametasone alla sera, mentre nei bambini con 0.25-0.5 mg/die. Questa dose può successivamente essere ridotta gradualmente a livelli che mantengano gli androgeni surrenalici nel range di normalità.
Il trattamento dell’ipertensione viene fatto utilizzando antagonisti dell’aldosterone, in quanto queste molecole esercitano anche altre azioni, quali risparmio di potassio e effetti anti-androgeni, di cui questi pazienti possono beneficiare. Non è consigliabile utilizzare tiazidi e diuretici dell’ansa.
Bibliografia
- Chrousos GP, Vingerhoeds A, Brandon D, et al. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest 1982, 69: 1261–9.
- Kino T, Vottero A, Charmandari E, Chrousos GP. Familial/sporadic glucocorticoid resistance syndrome and hypertension. Ann N Y Acad Sci 2002, 970: 101-11.
- Charmandari E, Kino T, Chrousos GP. Primary generalized familial and sporadic glucocorticoid resistance (Chrousos syndrome) and hypersensitivity. Endocr Dev 2013, 24: 67–85.
Resistenza al GH (sindrome di Laron)
Giorgio Radetti & Silvia Longhi
Reparto di Pediatria, Ospedale Regionale di Bolzano
Introduzione
La sindrome di Laron o sindrome da insensibilità all'ormone della crescita (GH) è stata descritta per la prima volta dal Professor Zvi Laron nel 1966, anno in cui riportò il caso di tre bambini affetti da bassa statura estrema, nati da una coppia di ebrei consanguinei provenienti dallo Yemen.
Si tratta di una malattia ereditaria, caratterizzata da un difetto genico del recettore del GH (GH-R), che viene trasmessa in modo autosomico recessivo, da cui l’importanza di proporre una consulenza genetica ai genitori di individui affetti, prima di una successiva gravidanza.
Finora sono stati descritti più di 250 casi al mondo, ma l’esatta incidenza non è ancora nota. La frequenza della s. di Laron varia da 1 a 9 casi su 1.000.000 ed è più elevata in popolazioni originarie di paesi del Vicino Oriente, come Israele, Arabia Saudita, Egitto e Iraq. Inoltre, sono stati rinvenuti molti casi in alcuni villaggi dell’Ecuador, in particolare nella regione di Loja, i cui abitanti hanno origini sefardite (spagnoli di origine ebrea).
La malattia sembra colpire indifferentemente i due sessi, anche se nella popolazione ecuadoregna le persone di sesso femminile sembrano maggiormente affette (F19:M2).
Patogenesi
Il GH è una proteina secreta dalle cellule acidofile dell'ipofisi anteriore, la quale promuove la crescita dei tessuti molli e scheletrici, mediante l’azione del proprio mediatore, l’insulin-like growth factor-I (IGF-I). Il GH stimola la secrezione dell’IGF-I principalmente a livello epatico, ma anche in altri tessuti periferici, mediante l’azione esercitata sul suo recettore.
Il GH-R è una proteina di 70 kD, appartenente alla superfamiglia delle citochine/ematopoietine, composta da una parte extra-cellulare coinvolta nel legame con il GH, da una parte trans-membrana e da una parte intra-cellulare implicata nella trasduzione del segnale. Le GH-BP circolanti rappresentano la parte extra-cellulare del recettore stesso. Una singola molecola di GH si lega a due recettori e induce una modificazione strutturale del recettore stesso. Di seguito, a cascata, si assiste a un’attivazione della tirosin-chinasi JAK2 e quindi all’attivazione di 4 diverse proteine STAT (STAT1, STAT3, STAT5a e 5b), della via PI3K e della via MAPK.
La STAT5b, la più importante nel processo di signalling, viene fosforilata da JAK2 e traslocata nel nucleo, dove regola la trascrizione dei geni che codificano per le proteine IGF-I, IGF-BP3 e ALS. L'IGF-I così prodotta viene rilasciata in circolo, dove si trova in un complesso ternario, associata a IGF-BP3 e ALS.
L’azione periferica della IGF-I è mediata dal legame con il suo recettore (IGF-IR) che, tramite il reclutamento di componenti citoplasmatici della via di trasduzione, conduce alla proliferazione cellulare e ad altri effetti metabolici.
La sindrome da insensibilità all'ormone della crescita è dovuta a una mutazione del gene che codifica per il GH-R.
Caratteristiche cliniche
La crescita intra-uterina e la lunghezza alla nascita sono spesso normali, mentre la crescita post-natale è generalmente rallentata, con età ossea ritardata e ridotta velocità di crescita. L’altezza finale dei soggetti non trattati è estremamente bassa: tra i 108-136 cm nelle femmine e 119-142 cm nei maschi (da -3 a -12 SD).
Lo sviluppo motorio è anch’esso ritardato, conseguentemente alla ridotta massa muscolare.
I neonati spesso presentano crisi ipoglicemiche e micropene e successivamente possono avere pubertà ritardata.
Il fenotipo è caratteristico con bozze frontali prominenti, ridotta circonferenza cranica, naso a sella, ipoplasia delle ossa facciali, sguardo a “sole calante”, capelli radi e sottili, acromicria, genitali piccoli e talvolta sclere blu (figura).

È di solito presente alterata composizione corporea, con osteopenia e obesità.
Storicamente questi pazienti erano considerati a rischio per insulino-resistenza e obesità, tendenza ora scomparsa da quando vengono trattati con IGF-I ricombinante.
Lo sviluppo intellettuale è di solito normale o solo modicamente ritardato.
Un fatto positivo è la bassa incidenza di cancro in questi pazienti, considerata secondaria ai bassi livelli circolanti di IGF-I.
Diagnosi
La diagnosi si basa sul riscontro dei classici segni clinici, associati a caratteristiche di laboratorio suggestive di resistenza all’ormone della crescita, quali elevate concentrazioni di GH (diretta conseguenza del mancato feed-back negativo da parte dell’IGF-I), bassi valori serici di GH-binding protein e livelli molto bassi di IGF-I.
La conferma dell’insensibilità periferica al GH si ottiene mediante il test di generazione di IGF-I, in cui non si osserva alcuna risposta di IGF-I dopo somministrazione di GH esogeno. Nella pratica si somministra GH alla dose di 0.025 mg/kg per 4 giorni consecutivi, con prelievi per IGF-I il primo (base) ed il 5° giorno: si definisce normale un incremento di IGF-I > 100 ng/mL.
La conferma molecolare si basa sul riscontro di mutazioni del gene codificante il GH-R. Attualmente sono state descritte più di 70 differenti mutazioni, in omozigosi o in eterozigosi composta, a carico del recettore del GH. La maggior parte di queste mutazioni sono a carico della parte extra-cellulare, deputata al legame con il GH, ma sono state descritte anche alterazioni della dimerizzazione del GH-R, come pure difetti della parte trans-membrana, che alterano l’ancoraggio del GH-R alla membrana cellulare, e alterazioni di tipo post-recettoriale (STAT5b), le quali in aggiunta all’insensibilità al GH sono pure causa di deficit immunitario, con infezioni polmonari ricorrenti e quadri di fibrosi polmonare.
Trattamento
Il trattamento si basa sulla somministrazione per via sottocutanea di mecasermina, IGF-I ricombinante, terapia che venne descritta alla fine degli anni ‘80 e che nel 2005 venne approvata dalla FDA e successivamente nel 2007 dall’EMEA.
Il trattamento con IGF-I, che non è raccomandato nei bambini di età < 2 anni per mancanza di dati su sicurezza ed efficacia, rappresenta il farmaco di scelta non solo nei pazienti con sindrome di Laron, ma anche nei casi di mancata produzione o resistenza all’IGF-I.
Il prodotto va somministrato per iniezione sottocutanea 2 volte al giorno prima dei pasti. La dose raccomandata iniziale è di 0.04 mg/kg per due volte al giorno, da aumentare gradualmente fino a raggiungere la dose massima di 0.12 mg/kg x 2/die.
Il successo del trattamento deve essere valutato sulla base della velocità di crescita. L’effetto del trattamento con IGF-I sulla crescita staturale nel primo anno di terapia oscilla tra 8-9 cm/anno, mentre negli anni successivi decresce fino a raggiungere i 4-5 cm/anno. La risposta è variabile e dipende dal grado del ritardo di crescita e dall’età al momento dell’inizio terapia. In ogni caso i risultati raggiunti sono significativamente inferiori a quelli osservati nei pazienti deficitari di ormone della crescita (GHD) trattati con GH.
Bibliografia
- David A, Hwa V, Metherell LA, et al. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev 2011, 32: 472-97.
- Savage MO, Burren CP, Rosenfeld RG. The continuum of growth hormone-IGF-I axis defects causing short stature: diagnostic and therapeutic challenges. Clin Endocrinol (Oxf) 2010, 72: 721-8.
- Buckway CK, Guevara-Aguirre J, Pratt KL, et al. The IGF-I generation test revisited: a marker of gh sensitivity. J Clin Endocrinol Metab 2001, 86: 5176-83.
Scheda IGF-I ricombinante
Giorgio Radetti & Silvia Longhi
Reparto di Pediatria, Ospedale Regionale di Bolzano
Meccanismo d’azione
IGF-I ricombinante che si lega al recettore naturale.
Indicazioni
Pazienti con sindrome di Laron, casi di mancata produzione o resistenza all’IGF-I.
Controindicazioni
Neoplasia attiva.
Non è raccomandato nei bambini di età < 2 anni per mancanza di dati su sicurezza ed efficacia.
Preparati e dosaggi
Mecasermina (Increlex) è disponibile in fl da 4 mL, contenenti 10 mg/mL (40 mg/fl), da conservare in frigorifero.
Va somministrato per iniezione sottocutanea 2 volte al giorno prima dei pasti. La dose raccomandata iniziale è di 0.04 mg/kg per due volte al giorno, da aumentare gradualmente fino a raggiungere la dose massima di 0.12 mg/kg x 2/die.
Effetti collaterali e precauzioni
Gli effetti collaterali più comuni sono rappresentati dall’ipoglicemia, per cui viene consigliato di somministrare il farmaco prima dei pasti e di eseguire monitoraggio glicemico pre- e post-prandiale, soprattutto all’inizio della terapia.
Occasionalmente è stata descritta ipertrofia nel sito di iniezione e ipertrofia tonsillare. A causa della possibilità di ipertrofia del tessuto linfoide, si consiglia una valutazione otorinolaringoiatrica nel caso insorgano apnea nel sonno, otiti sierose croniche dell’orecchio medio, russamento, ecc.
Un esame del fondo dell’occhio deve essere eseguito qualora compaiano sintomi clinici ricollegabili a ipertensione endocranica, quali cefalea persistente, nausea o vomito (pseudotumor cerebri).
Nei pazienti che manifestano rapida crescita possono verificarsi epifisiolisi e progressione della scoliosi.
Limitazioni prescrittive
Attualmente il farmaco non prevede piano terapeutico nè nota AIFA, ma è un farmaco ospedaliero e per tale motivo è vendibile al pubblico su prescrizione di centri ospedalieri o di specialisti secondo art. 93 DL 219/06.
Altre resistenze
Marco Bonomi
Divisione Endocrinologia e Metabolismo, Istituto Auxologico Italiano, Milano
Resistenza al TRH
La sindrome da resistenza al TRH è una forma particolare di ipotiroidismo centrale congenito isolato, dovuta a una ridotta o assente sensibilità delle cellule ipofisarie tireotrope allo stimolo del TRH. La causa molecolare risiede in un’alterazione a carico del gene del recettore del TRH (TRHR), che può comportare la mancata produzione della proteina matura o la produzione di una proteina con un’alterata capacità di legame dell’ormone o di attivazione della via intra-cellulare dei secondi messaggeri (1,2). È un disordine recessivo, che si manifesta clinicamente solo in presenza di un difetto genico in omozigosi o in eterozigosi composita, mentre i soggetti eterozigoti risultano non affetti.
Il quadro clinico alla nascita è in genere non evidente, al contrario di quanto avviene nei casi d’ipotiroidismo centrale congenito da mutazioni del gene che codifica per la ß-subunità del TSH (TSHß) (3) e che sono drammaticamente manifesti fin dai primi giorni di vita. Questa differenza potrebbe ricondursi all’attività costitutiva del recettore del TSH (TSHR), che permette una certa stimolazione autonoma della tiroide e una minima produzione ormonale pur in assenza di un adeguato stimolo da parte della tireotropina ipofisaria. Il quadro clinico di queste forme d’ipotiroidismo centrale congenito isolato da resistenza al TRH diviene invece manifesto in epoca post-natale, per la presenza di un ritardo di crescita staturale e talora una ridotta performance scolastica. Si associano anche segni e sintomi comuni agli stati di ipotiroidismo primario (facile affaticabilità, sonnolenza, ridotta capacità di concentrazione, ecc), che possono però essere sfumati e passare inosservati come nel caso estremo della probanda che abbiamo recentemente descritto e che giungeva alla diagnosi in età adulta pur essendo portatrice di una resistenza completa al TRH (2).
La diagnosi di resistenza al TRH si basa sul riscontro biochimico di bassi livelli di FT4 in associazione a livelli di TSH inappropriatamente normali. I soggetti affetti possono pertanto sfuggire ai metodi di screening neonatale per ipotiroidismo congenito se si basano sulla determinazione del solo TSH. Caratteristicamente, a seguito di stimolazione esogena con TRH, questi pazienti presentano un’assente o alterata risposta sia del TSH che della PRL. Non sono presenti altri difetti ormonali ipofisari associati e il quadro neuroradiologico è privo di alterazioni a carico della regione ipotalamo-ipofisaria. La presenza, in questi pazienti, di valori di FT4 bassi associati a valori di TSH dentro gli intervalli di normalità suggerisce, come per altre forme d’ipotiroidismo centrale (4), che queste molecole di TSH circolante possano avere una ridotta attività biologica, probabilmente legata a un alterato processo di glicosilazione post-traduzionale. L’analisi genetica del gene TRHR conferma il sospetto diagnostico.
La terapia di queste forme d’ipotiroidismo centrale si basa sulla terapia sostitutiva con levo-tiroxina sodica, il cui monitoraggio andrà impostato sui livelli circolanti di ormoni tiroidei liberi (3).
Recentemente è stato descritto un nuovo meccanismo molecolare che porta a un certo grado di resistenza al TRH, legato a varianti del gene IGSF1 che mappa sul cromosoma X (5). Benchè la fisiopatologia di queste forme d’ipotiroidismo centrale idiopatico X-linked non sia ancora del tutto chiarita, i pazienti maschi, portatori in emizigosi di queste varianti, presentano un quadro biochimico con bassi livelli di TSH, spesso associati a deficit di PRL, e una risposta al TRH test che risulta deficitaria in età infantile e ai limiti della normalità in età adulta (6). Il quadro clinico di questi pazienti associa all’ipotiroidismo centrale congenito anche la presenza di macro-orchidismo e uno sviluppo puberale disarmonico con, talora, aumento dell’indice di massa corporea e sindrome metabolica (5,6).
Resistenza al TSH
È una condizione caratterizzata da un’alterata sensibilità delle cellule tiroidee all’azione della tireotropina. Questa condizione è generalmente associata ad alterazioni del recettore del TSH (TSHR), che causano quadri clinici eterogenei in dipendenza del tipo di compromissione recettoriale. Possiamo avere quadri clinici di resistenza completa o parziale di grado moderato, che sono legati a una trasmissione di tipo autosomico recessivo e che pertanto colpiscono entrambi gli alleli del gene TSH (in omozigosi o in eterozigosi composita), oppure forme parziali di grado lieve che sono invece caratterizzate da una modalità di trasmissione autosomica dominante e che pertanto sono presenti in eterozigosi semplice (7). In quest’ultimo caso è stato dimostrato che il recettore mutato esercita un effetto di dominanza negativa sul recettore wild-type, attraverso il meccanismo dell’oligomerizzazione tipica di questi recettori, riducendone la funzionalità (8). Più raramente la resistenza al TSH può essere invece legata a mutazioni inattivanti a carico del gene GNAS, che codifica per la subunità alfa della proteina G stimolatoria e che causa anche lo pseudoipoparatiroidismo (PHP) di tipo Ia. Questi pazienti presentano alti livelli di TSH con livelli di ormoni tiroidei normali o ridotti.
Il quadro clinico dipende strettamente dal grado di resistenza recettoriale (4). Poiché il TSH costituisce il maggiore stimolo fisiologico della funzione e della proliferazione dei tireociti, nei quadri di profonda insensibilità al TSH dovuti a resistenza recettoriale completa, abbiamo quadri d’ipotiroidismo importante con ipoplasia ghiandolare e livelli di TSH marcatamente elevati. Questi casi sono tipicamente diagnosticati in epoca neonatale come forme d’ipotiroidismo congenito. Quando invece la refrattarietà al TSH è incompleta, l’elevazione del TSH può in parte compensare il difetto stimolatorio e i quadri clinici che ne derivano sono forme di ipotiroidismo lieve, con volumi ghiandolari tiroidei spesso nella norma o lievemente ridotti e livelli di ormoni tiroidei nella norma. Questo stesso quadro risulta ancora più sfumato, sia da un punto di vista clinico che ormonale, nei casi con mutazione in eterozigosi semplice del gene del TSHR.
Dal punto di vista diagnostico, la resistenza al TSH si caratterizza, quindi, per la presenza di TSH elevato in presenza di valori bassi o normali di FT4 e volume tiroideo normale o ridotto (4). Queste stesse caratteristiche cliniche e biochimiche possono essere riscontrate frequentemente nella popolazione, ma solo una minima parte di questi soggetti presenta una reale resistenza al TSH (4). Sarà quindi necessario escludere altre possibili cause, tra cui, in primis, la patologia autoimmune tiroidea, mediante valutazione della storia clinica del paziente, misurazione degli auto-anticorpi e valutazione del pattern ecografico della ghiandola tiroidea. Altre possibili cause possono essere i difetti dell’attività biologica del TSH o altre forme di ipotiroidismo primario congenito, tra cui quelle forme dovute ad alterazione dei fattori di trascrizione tiroidei.
I pazienti con le forme complete di resistenza al TSH vanno trattati con terapia sostitutiva con levo-tiroxina sodica, mentre molte evidenze dimostrano che le forme incomplete sono compensate dall’aumento del TSH. In questi casi, in cui l’aumento del TSH porta a normali livelli circolanti di ormoni tiroidei, rimane tutt’oggi discusso se sia necessario intraprendere un trattamento sostitutivo (9).
Resistenza al GnRH
È una malattia rara a trasmissione autosomico recessiva, legata alla presenza di varianti genetiche in omozigosi o in eterozigosi composita a carico del gene del recettore del GnRH, GNRHR (10). Tali varianti compromettono il funzionamento recettoriale a livello della sua capacità di legare l’ormone e/o di trasdurre il segnale all’interno della cellula. Ne consegue un difetto d’azione dell’ormone GnRH e una mancata attivazione dell’asse ipotalamo-ipofisi-gonadi (HPG).
Clinicamente abbiamo un quadro d’ipogonadismo ipogonadotropo isolato, la cui entità dipende dal grado, totale o parziale, di mancanza di produzione di ormoni sessuali (10). Nei casi più severi e limitatamente ai soggetti maschi si possono avere segni alla nascita quali criptorchidismo, micropene e/o ipospadia. Tali segni sono indicativi dell’alterata attivazione dell’asse HPG già durante la vita fetale. In altri casi di minore severità i sintomi si manifestano in epoca peri-puberale, con ritardo o assente sviluppo puberale e proporzioni eunucoidi nei maschi, amenorrea primaria e ritardo di crescita nelle femmine. Nei casi più lievi il difetto può anche divenire evidente in epoca post-puberale o in età adulta, con sintomi quali calo della libido, infertilità, osteoporosi, astenia, depressione, facile affaticabilità, anemia e deficit erettile nel maschio e amenorrea nella femmina.
La diagnosi è confermata dalla presenza di valori di gonadotropine bassi o inappropriatamente normali, con livelli di steroidi sessuali bassi (11). Il test di stimolo con GnRH non mostra alcuna risposta delle gonadotropine. È necessario eseguire una risonanza magnetica nucleare della regione sellare, per escludere alterazioni ipotalamo-ipofisarie che possano giustificare il difetto ormonale (11). È sempre necessario valutare anche la funzione olfattiva, per escludere che si tratti di una forma di sindrome di Kallmann (11). La conferma diagnostica di questa resistenza viene dall’analisi genetica del gene GnRHR (10).
Il trattamento consiste nella terapia sostitutiva con testosterone o estro-progestinici secondo il sesso del/della paziente e, in caso di desiderio di fertilità si procede alla stimolazione delle gonadi mediante la somministrazione di gonadotropine esogene (11).
Resistenza alle gonadotropine
Le gonadotropine ipofisarie, LH e FSH, sono essenziali per il corretto sviluppo puberale e per la successiva funzione riproduttiva. LH e FSH esercitano la loro funzione attraverso specifici recettori di membrana, LHR e FSHR, detti recettori degli ormoni glicoproteici (GpHRs), appartenenti alla superfamiglia dei recettori accoppiati a proteine G (GPCRs) (12). L’LH di secrezione ipofisaria e la Gonadotropina Corionica, CG, di origine placentare, condividono il medesimo recettore, LH/CGR. Le mutazioni inattivanti a carico dei geni LH/CGR e FSHR sono alla base delle forme di resistenza alle gonadotropine e rappresentano un importante modello naturale per la comprensione delle differenti funzioni di LH e FSH sulle gonadi nei soggetti umani (11,13).
Mutazioni inattivanti, in omozigosi o in eterozigosi composita, a carico di LH/CGR causano, nei soggetti di sesso maschile, uno spettro fenotipico che dipende dalla gravità di compromissione della funzione recettoriale (11,13). Nei casi più gravi, a causa dell’importante ipoplasia delle cellule di Leydig e del conseguente deficit androgenico presente già durante la vita fetale, possiamo avere ambiguità o chiara femminilizzazione dei genitali alla nascita, con mancata fusione dei lembi scrotali a mimare una vagina a fondo cieco, criptorchidismo a mimare la presenza di ovaie, micropene a mimare un processo clitorideo. Questi soggetti vengono spesso scambiati per soggetti di sesso femminile e quindi riconosciuti solo in epoca puberale per mancato sviluppo mammario e ipotetica amenorrea primaria. Nei casi meno gravi, in cui viene mantenuta una certa risposta di LH/CGR al suo ligando, il fenotipo può variare da un’incompleta differenziazione maschile con micropene e/o ipospadia, a un ritardo di sviluppo puberale ma senza ambiguità genitale. È stata inoltre descritta una mutazione particolare del gene LH/CGR, che consiste nella delezione completa dell’esone 10 del gene, che si associa a un normale fenotipo maschile alla nascita ma assente sviluppo puberale (14). Questo fenotipo suggerisce che questo recettore LH/CGR mutante è in grado di rispondere allo stimolo di hCG fetale, mentre è resistente all’LH ipofisario.
Anche nei soggetti di sesso femminile le mutazioni inattivanti del gene LH/CGR portano a quadri fenotipici variabili, in accordo con il tipo di mutazione genica e con la compromissione del recettore (11,13). Possiamo avere quadri di amenorrea primaria o quadri meno gravi con oligomenorrea, sempre associati con una costante infertilità. I livelli di estradiolo e di progesterone restano costantemente bassi come normalmente nella fase follicolare, mentre le ovaie sono di volume regolare o addirittura ingrandite con cisti. In entrambi i sessi, i livelli di LH sono aumentati con steroidi sessuali ridotti, mentre i livelli di FSH restano all’interno dell’intervallo di normalità.
Le mutazioni inattivanti del gene del recettore di FSH, FSHR, sono molto più rare di quelle di LH/CGR (11,13). Tali mutazioni, nel sesso femminile, si associano a un’insufficienza ovarica prematura parziale o completa, mentre, nel sesso maschile, portano a una variabile compromissione della spermatogenesi con ridotto volume testicolare.
Resistenza agli estrogeni
È una malattia estremamente rara, geneticamente determinata e con ereditarietà autosomica recessiva. I soggetti affetti presentano una variante biallelica a carico del gene ESR1, che codifica per il recettore nucleare degli estrogeni, con conseguente insensibilità all’ormone (15,16).
Il quadro clinico si diversifica a seconda del sesso del paziente. Nel paziente di sesso maschile descritto in letteratura (15,17,18) si è osservata una crescita lineare che continuava anche in età adulta, con l’evidenza di un’incompleta saldatura delle epifisi all’esame radiografico. Il paziente presentava normale sviluppo puberale e mascolinizzazione. Erano invece presenti ridotta mineralizzazione ossea e alterato profilo lipidico, con evidenza di aterosclerosi precoce, alterata tolleranza glucidica e iperinsulinemia.
Il soggetto di sesso femminile descritto in letteratura (16) affetto da resistenza agli estrogeni presentava amenorrea primaria, assente sviluppo mammario ma normale sviluppo di peluria pubica, ridotta età ossea, utero ipoplasico e ovaie ingrandite e policistiche. La paziente presentava inoltre un assente spurt di crescita puberale. Non era presente un’alterata tolleranza glucidica.
Dal punto di vista ormonale, questi pazienti presentano livelli di estrogeni marcatamente elevati con, nel soggetto maschile, normali livelli di testosterone e livelli elevati di LH e FSH.
Resistenza all’ACTH
È una malattia rara ed eterogenea, geneticamente determinata, che appartiene alle forme familiari di deficit di glucocorticoidi (FDG) (19,20). Nella FDG da resistenza all’ACTH le cellule della zona fascicolata della corteccia surrenalica non sono in grado di rispondere correttamente allo stimolo dell’ACTH e, di conseguenza, non producono cortisolo.
Abbiamo quindi quadri clinici da iposurrenalismo, con livelli di cortisolo bassi o addirittura indosabili e livelli di ACTH estremamente elevati. La mancanza di cortisolo porta a ipoglicemia e/o incapacità di nutrirsi nei neonati o nella prima infanzia, mentre può manifestarsi con infezioni ricorrenti o crisi epilettiche ipoglicemiche nel bambino (21). Se non viene prontamente riconosciuta e diagnosticata, questa forma particolare di iposurrenalismo può portare a difficoltà d’apprendimento secondario, ipoglicemie ricorrenti, ipotensione e sintomi neurologici ed è potenzialmente fatale. L’eccesso di ACTH risulta spesso in un’iperpigmentazione cutanea e delle mucose da iperstimolazione del recettore 1 delle melanocortine (MC1R).
La causa genetica di queste forme di FDG da resistenza all’ACTH è rappresentata da mutazioni a carico del gene per ACTH recettore (ACTHR) o recettore 2 delle melanocortine (MC2R), che porta alla FDG di tipo 1, o da mutazioni a carico della proteina accessoria del recettore 2 delle melanocortine, MRAP, che porta alla FDG di tipo 2 (19,20). Il MC2R è un recettore di membrana accoppiato a proteine G, che viene attivato dal legame di ACTH, mentre MRAP è una piccola proteina accessoria trans-membranaria, che facilita il trasporto di MC2R dal reticolo endoplasmico alla membrana plasmatica. Ne consegue che queste due forme di FDG sono dovute a un difetto d’azione di ACTH che può essere dovuto a un legame difettivo di ACTH al suo recettore (FDG tipo 1) o a un mancato trasporto di ACTHR in membrana (FDG di tipo 2).
Altre forme ancora più rare e particolari di resistenza all’ACTH sono dovute a mutazioni del gene STAR (steroidogenic acute regulatory protein), che causano un inefficiente trasporto di colesterolo attraverso la membrana mitocondriale con un conseguente difetto nella via steroidogenica, o, come più recentemente descritto, a mutazioni dei geni MCM4 (mini-chromosome maintenance-deficient 4 homologue) e NNT (nicotinamide nucleotide transhydrogenase) (19,20). Il prodotto del gene MCM4 è importante per la replicazione del DNA e per la stabilità del genoma, mentre il prodotto del gene NNT è implicato nel sistema di glutatione-reduttasi che protegge le cellule dai radicali liberi dell’ossigeno. Il corretto funzionamento di questi due geni protegge la corteccia surrenalica da stress replicativi e ossidativi, garantendone una corretta funzionalità e sensibilità allo stimolo endogeno con ACTH.
Il trattamento di queste forme di resistenza ad ACTH consiste nella terapia sostitutiva con glucocorticoidi.
Resistenza all’insulina
L’insulino-resistenza è caratterizzata da una ridotta sensibilità dei tessuti all’azione dell’insulina, causata da un’alterata funzione recettoriale (22). Dal punto di vista biologico il problema può localizzarsi a diversi livelli rispetto al recettore insulinico, comprese le varie possibili sovrapposizioni:
- le forme pre-recettoriali sono dovute a processi (ad esempio anticorpi anti-insulina) che impediscono un pieno legame dell’ormone al recettore;
- le forme recettoriali sono tipicamente dovute a varianti del recettore insulinico, come il leprecaunismo (sindrome di Donohue) o nelle sindromi da Achantosis Nigricans (sindrome di Rabson-Mendenhall);
- le forme post-recettoriali sono sicuramente le più diffuse e sono raramente associate a riduzione del numero dei recettori, d’affinità di legame o d’attivazione recettoriale (22).
Le cause della resistenza insulinica possono essere molteplici: ormonali (difetto qualitativo dell’insulina secreta o eccessiva sintesi di ormoni ad azione contro-insulare) o genetiche (varianti recettoriali). Nella maggior parte dei casi, tuttavia, le cause d’insulino-resistenza non sono chiaramente identificabili. Dal punto di vista fisiopatologico, l’insulino-resistenza viene normalmente compensata da un’aumentata secrezione di insulina da parte del pancreas (22). Tuttavia, questo meccanismo compensatorio può venir meno nel tempo, dando luogo alla manifestazione di un quadro di diabete. Va inoltre tenuto presente che, anche quando gli individui riescono a mantenere una normale glicemia a spese di un’aumentata secrezione insulinica, essi hanno un’elevata probabilità di sviluppare una serie di fattori di rischio raggruppati in quella che è comunemente definita la sindrome da insulino-resistenza (23). È, infatti, noto che l’insulino-resistenza e l’iperinsulinismo compensatorio predispongono al rischio di sviluppare diverse patologie, tra cui il diabete mellito di tipo 2, l’ipertensione arteriosa e l’aterosclerosi con la malattia cardiovascolare (22,24).
L’individuazione dei soggetti insulino-resistenti non è facile, poiché non è disponibile un test clinico che permetta di valutare direttamente in vivo l’azione insulinica. L’iperinsulinemia o la misurazione di altri surrogati dell’insulino-resistenza (ad esempio l’andamento dei livelli di glicemia dopo test da carico orale con glucosio) possono essere strumenti utili e hanno un certo grado di affidabilità, ma sono comunque test indiretti.
Il trattamento più efficace per l'insulino-resistenza è dato dalla pratica di regolare attività fisica, associata al dimagrimento e all'adozione di una dieta basata sulla moderazione calorica e sul consumo di alimenti a basso indice glicemico. Possono essere utili anche i presidi in grado di ridurre o rallentare l'assorbimento intestinale degli zuccheri (es. acarbosio, integratori di fibra). Alcuni farmaci come la metformina si sono dimostrati efficaci anche nel trattamento dell'insulino-resistenza; tuttavia è molto importante intervenire prima di tutto sulla dieta e sul livello di attività fisica, ricorrendo alla terapia farmacologica solo quando le modifiche dello stile di vita non sono sufficienti.
Resistenza alla vitamina D
È alla base di una forma genetica autosomica recessiva di rachitismo, che prende il nome di rachitismo ereditario vitamina D-resistente (HVDRR) (25). La causa di questa forma di rachitismo è legata a mutazioni inattivanti del gene che codifica per il recettore della vitamina D, VDR.
La HVDRR è caratterizzata da ipocalcemia, ipofosfatemia e iperparatiroidismo compensatorio, con rachitismo grave a insorgenza infantile (25). I bambini affetti possono presentare anche alopecia del cuoio capelluto e dell’intero corpo.
I bambini di solito non rispondono al trattamento con calcitriolo, infatti, i loro livelli endogeni sono spesso molto elevati. Il trattamento di successo richiede un'inversione dell’ipocalcemia e dell’iperparatiroidismo secondario e di solito consiste nella somministrazione di alte dosi di calcio per via orale o, a volte, per via endovenosa, al fine di bypassare il difetto intestinale del VDR (25).
Bibliografia
- Collu R, Tang J, Castagné J, et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab 1997, 82: 1561-5.
- Bonomi M, Busnelli M, Beck-Peccoz P, et al. A family with complete resistance to thyrotropin-releasing hormone. N Engl J Med 2009, 360: 731-4.
- Persani L. Central hypothyroidism: pathogenic, diagnostic, and therapeutic challenges. J Clin Endocrinol Metab 2012, 97: 3068-78.
- Beck-Peccoz P, Persani L, Calebiro D, et al. Syndromes of hormone resistance in the hypothalamic-pituitary-thyroid axis. Best Pract Res Clin Endocrinol Metab 2006, 20: 529-46.
- Sun Y, Bak B, Schoenmakers N, et al. Loss-of-function mutations in IGSF1 cause an X-linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet 2012, 44: 1375-81.
- Joustra SD, Schoenmakers N, Persani L, et al. The IGSF1 deficiency syndrome: characteristics of male and female patients. J Clin Endocrinol Metab 2013, 98: 4942-52.
- Alberti L, Proverbio MC, Costagliola S, et al. Germline mutations of TSH receptor gene as cause of nonautoimmune subclinical hypothyroidism. J Clin Endocrinol Metab 2002, 87: 2549-55.
- Calebiro D, de Filippis T, Lucchi S, et al. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum Mol Genet 2005, 14: 2991-3002.
- Calebiro D, Gelmini G, Cordella D, et al. Frequent TSH receptor genetic alterations with variable signaling impairment in a large series of children with nonautoimmune isolated hyperthyrotropinemia. J Clin Endocrinol Metab 2012, 97: E156-60.
- Bonomi M, Libri DV, Guizzardi F, et al; Idiopathic Central Hypogonadism Study Group of the Italian Societies of Endocrinology and Pediatric Endocrinology and Diabetes. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl 2012, 14: 49-56.
- Latronico AC, Arnhold IJ. Gonadotropin resistance. Endocr Dev 2013, 24: 25-32.
- Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev 2002, 23: 141-74.
- Huhtaniemi I, Alevizaki M. Gonadotrophin resistance. Best Pract Res Clin Endocrinol Metab 2006, 20: 561-76.
- Gromoll J, Eiholzer U, Nieschlag E, Simoni M. Male hypogonadism caused by homozygous deletion of exon 10 of the luteinizing hormone (LH) receptor: differential action of human chorionic gonadotropin and LH. J Clin Endocrinol Metab 2000, 85: 2281-6.
- Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med 1994, 331: 1056-61.
- Quaynor SD, Stradtman EW Jr, Kim HG, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med 2013, 369: 164-71.
- Sudhir K, Chou TM, Messina LM, et al. Endothelial dysfunction in a man with disruptive mutation in oestrogen-receptor gene. Lancet 1997, 349: 1146-7.
- Smith EP, Specker B, Bachrach BE, et al. Impact on bone of an estrogen receptor-alpha gene loss of function mutation. J Clin Endocrinol Metab 2008, 93: 3088-96.
- Meimaridou E, Hughes CR, Kowalczyk J, et al. ACTH resistance: genes and mechanisms. Endocr Dev 2013, 24: 57-66.
- Meimaridou E, Hughes CR, Kowalczyk J, et al. Familial glucocorticoid deficiency: new genes and mechanisms. Mol Cell Endocrinol 2013, 371: 195-200.
- Cooray SN, Chan L, Metherell L, et al. Adrenocorticotropin resistance syndromes. Endocr Dev 2008, 13: 99-116.
- Paneni F, Costantino S, Cosentino F. Insulin resistance, diabetes, and cardiovascular risk. Curr Atheroscler Rep 2014, 16: 419.
- Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37: 1595-607.
- Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med 2014, 371: 2237-8.
- Feldman D, J Malloy P. Mutations in the vitamin D receptor and hereditary vitamin D-resistant rickets. Bonekey Rep 2014, 3: 510.
Patologie endocrine in età pediatrica
Ipopituitarismi congeniti
Carla Bizzarri
UOC Endocrinologia e Diabetologia, Ospedale Bambino Gesù, IRCCS, Roma
(aggiornato al 10 ottobre 2016)
INTRODUZIONE
L’ipofisi è costituita dall’adeno-ipofisi, che comprende i lobi anteriore e intermedio (derivati dall'ectoderma orale), e dalla neuro-ipofisi (derivata dall’ectoderma neurale).
Il lobo anteriore contiene cinque tipi di cellule (con i rispettivi ormoni increti): somatotrope (GH), tireotrope (TSH), lattotrope (PRL), gonadotrope (FSH e LH) e corticotrope (ACTH). Il lobo intermedio contiene le melanotrope, secernenti pro-opiomelanocortina, il precursore di MSH ed endorfine. Il lobo posteriore è composto dalle proiezioni assonali di neuroni, i cui corpi cellulari risiedono nei nuclei ipotalamici sopra-ottico e para-ventricolare, secernenti rispettivamente AVP e ossitocina.
Lo sviluppo dell’ipofisi dipende dall’espressione sequenziale, in senso sia temporale che spaziale, di fattori di trascrizione e molecole di segnale. L’ipopituitarismo congenito può essere causato da mutazioni di uno qualsiasi dei geni coinvolti nello sviluppo dell'ipofisi (tab 1).
| Tabella 1 Caratteristiche cliniche dell’ipopituitarismo congenito legato a mutazioni dei principali geni codificanti per i fattori di trascrizione coinvolti nello sviluppo dell’ipofisi (adattata da 2) |
||||
| Fattore di trascrizione | Trasmissione | Difetti ormonali | Quadro RM | Altre caratteristiche cliniche |
| POU1F1 (PIT1) | AR, AD | GH, TSH, PRL | APH | |
| PROP1 | AR | GH, TSH, LH, FSH, PRL; deficit di ACTH a presentazione tardiva | APH, N, E | Talvolta transitoria iperplasia dell’ipofisi anteriore |
| HESX1 | AR, AD | IGHD, CPHD | APH, EPP, ACC | Displasia setto-ottica |
| LHX3 | AR | GH, TSH, LH, FSH, PRL (talvolta ACTH) | APH, N, E | Limitata mobilità del collo, colonna cervicale corta, sordità neuro-sensoriale |
| LHX4 | AD | CPHD (GH, TSH, ACTH, talvolta FSH-LH) | APH, EPP | Anomalie cerebellari |
| SOX2 | AD (de novo) | Ipogonadismo centrale, talvolta GHD | APH | Anoftalmia/microftalmia, atresia esofagea, anomalie genitali, amartoma ipotalamico, sordità neuro-sensoriale, diplegia |
| SOX3 | XL | IGHD o CPHD | APH, EPP | Ritardo mentale |
| OTX2 | AD | IGHD o CPHD (GH, TSH, PRL, LH, FSH) | N, APH, EPP | Anoftalmia/microftalmia bilaterale |
| TBX19 (T-PIT) | AR | ACTH | N | Ipoglicemia neonatale grave |
| PC1 | AR | ACTH, LH, FSH | N | Ipoglicemia ricorrente, obesità |
| DAX-1 | XL | FSH, LH | N | Ipoplasia congenita del surrene, con insufficienza surrenalica neonatale o a esordio tardivo |
| AR = autosomica recessiva; AD = autosomica dominante; XL = X-linked; APH = ipoplasia dell’ipofisi anteriore; N = normale; E = iperplasia dell’ipofisi anteriore; EPP = ectopia della neuro-ipofisi; ACC = agenesia del corpo calloso | ||||
L’ipopituitarismo congenito si manifesta sia come deficit isolato di un ormone, più comunemente deficit di GH isolato (IGHD), sia come deficit combinato di più ormoni (CPHD). I deficit ormonali possono presentarsi come parte di una sindrome comprendente anomalie nelle strutture che condividono una comune origine embriologica, come l'occhio e il prosencefalo. Le mutazioni nei geni implicati nelle prime fasi di sviluppo dell'ipofisi tendono a provocare forme sindromiche di ipopituitarismo, associato a difetti extra-ipofisari e anomalie della linea mediana. Mutazioni di geni implicati nella differenziazione di particolari tipi di cellule o codificanti specifiche subunità ormonali danno luogo a carenze isolate di ormoni ipofisari.
MANIFESTAZIONI CLINICHE NEL PERIODO NEONATALE
I neonati con ipopituitarismo congenito hanno peso e lunghezza normali alla nascita, mentre è stata descritta un’aumentata prevalenza di asfissia perinatale.
Possono presentare sintomi non specifici, associati o meno ad anomalie delle strutture aventi origine embriologica comune con quella ipofisaria (occhi, setto pellucido, corpo calloso) e di altre strutture della linea mediana (labio-palatoschisi, anomalie genitali). In alternativa, possono essere inizialmente asintomatici e sviluppare difetti ormonali nel corso del tempo. Per questa ragione, nei neonati con ipoplasia dei nervi ottici, anomalie della linea mediana o sindromi note per essere associate all’ipopituitarismo è necessario un follow-up endocrino a lungo termine, anche se le indagini ormonali iniziali sono normali.
I sintomi sono spesso correlati alla presenza di ipoglicemia, con conseguenti segni di neuroglicopenia, quali letargia, crisi di apnea, irritabilità, convulsioni, scarso incremento ponderale. Possono associarsi iposodiemia non accompagnata da iperkaliemia, instabilità della temperatura corporea, sepsi ricorrenti, instabilità emodinamica e colestasi neonatale con ittero prolungato. Può essere presente nistagmo, correlato a ipoplasia dei nervi ottici o agenesia del corpo calloso.
L'ipoglicemia è dovuta principalmente alla mancanza di ACTH, in neonati con CPHD o con deficit isolato di ACTH; più raramente può essere associata a IGHD. Poiché i glucocorticoidi attivano il flusso biliare, la carenza di cortisolo può ritardare la maturazione fisiologica di sintesi e trasporto degli acidi biliari, con conseguente colestasi. L’iperbilirubinemia coniugata si manifesta a un'età media di 13 giorni ed è seguita da aumento delle transaminasi 2-4 settimane più tardi, mentre la gamma-GT rimane normale. Il deficit di TSH determina instabilità della temperatura corporea e contribuisce al prolungato ittero neonatale. Nei maschi, il deficit di gonadotropine è suggerito dalla presenza di micropene, associato o meno a criptorchidismo, in quanto la crescita del pene e la discesa del testicolo dipendono dalla normale secrezione fetale di LH e testosterone durante il II e il III trimestre di gravidanza.
ITER DIAGNOSTICO
La diagnosi di ipopituitarismo in epoca neonatale è difficile, per fattori come l'immaturità dell'asse ipotalamo-ipofisi, la mancanza di dati normativi appropriati per l’età e la controindicazione per alcuni test di stimolo. Sebbene fino al 52% dei neonati ipopituitarici possa avere complicazioni quali ipoglicemia, iponatremia e sepsi, la diagnosi viene posta nel periodo neonatale solo nel 23% dei casi.
Deficit di ACTH e deficit di GH
Poiché il deficit di ACTH e l’ipocortisolismo rappresentano un pericolo per la vita, nel sospetto di ipopituitarismo congenito è fondamentale indagare per prima la funzionalità dell’asse ipotalamo-ipofisi-surrene.
Nei primi sei mesi di vita il ritmo circadiano del cortisolo non è ancora presente. I risultati del cortisolo basale su campioni prelevati al mattino rispetto a quelli della sera non sono conclusivi e il concomitante trattamento con steroidi per eventuali patologie perinatali può complicarne l'interpretazione. Il test all’ipoglicemia insulinica è controindicato per tutto il primo anno di vita. Il test standard con ACTH è sicuro e semplice, ma ha una sensibilità dell’80%, di conseguenza può dare luogo a risultati falsi negativi. Il test con CRH consente la valutazione dell'asse nei neonati asintomatici, ma i dati di riferimento sono scarsi e il test è controindicato nei neonati malati o piccoli.
Nel lattante con ipoglicemia da sospetto ipopituitarismo (ad es. per la presenza di anomalie della linea mediana), gli esami ormonali in corso di ipoglicemia spontanea possono essere sufficienti a porre diagnosi: si considerano diagnostici GH < 5 ng/mL e cortisolemia < 10 µg/dL.
È importante sottolineare che:
- la glicemia nel neonato è normalmente più bassa che nell’adulto e la definizione dell’ipoglicemia in epoca neonatale prevede limiti di riferimento diversi (tab 2);
- i test di stimolo per GH sono controindicati nei bambini di età < 1 anno;
- livelli di IGF-1 bassi sono poco indicativi di GHD, perché fortemente influenzati dalle condizioni nutrizionali e da eventuali patologie concomitanti;
- l’IGF-binding protein-3 sembra risentire meno di questi fattori, ma il suo dosaggio non è sempre disponibile;
- in epoca neonatale i livelli di GH sono normalmente elevati, per cui valori random di GH basale < 7 ng/mL suggeriscono di per sé la diagnosi di GHD.
| Tabella 2 Cut-off per ipoglicemia nelle diverse età (in mg/dL) |
||
| Sangue intero | Plasma | |
| Neonato a termine | < 35 | < 40 |
| Pretermine | < 30 | < 35 |
| Lattante | < 45 | < 50 |
Deficit di TSH
L’ipotiroidismo centrale rappresenta il 13.5% dei casi di ipotiroidismo congenito permanente. La maggior parte dei pazienti con ipotiroidismo centrale (78%) ha ulteriori deficit di ormoni ipofisari. La diagnosi di deficit di TSH è suggerita da bassi livelli sierici di FT4 associati a livelli di TSH inappropriati (nel range della norma o lievemente aumentati). Il test con TRH non è necessario per la diagnosi di ipotiroidismo centrale.
Deficit di gonadotropine
Il neonato con micropene, associato o meno a criptorchidismo, è sospetto per essere affetto da ipogonadismo ipogonadotropo. Nel maschio, l'aumento post-natale di LH, FSH e testosterone raggiunge il suo picco tra 4 e 10 settimane (periodo definito della mini-pubertà) e poi declina dai 6 mesi di età. Nelle femmine l'aumento di FSH ed LH può essere rilevato fino a circa 2 anni di età. Esiste quindi una finestra di opportunità per la diagnosi precoce di ipogonadismo ipogonadotropo mediante la valutazione dei livelli basali di LH, FSH e steroidi gonadici, eventualmente associata a test di stimolo con GnRH e hCG. I neonati con ipogonadismo ipogonadotropo hanno basse concentrazioni basali di LH ed FSH e una ridotta risposta delle gonadotropine al GnRH. Il test all’hCG mostra invece una risposta normale del testosterone (> 100 ng/dL).
Diabete insipido
Il diabete insipido centrale può manifestarsi nei neonati con difetti della linea mediana, è invece assente nelle forme di ipopituitarismo da mutazioni dei fattori di trascrizione. Il neonato e il lattante con diabete insipido sono particolarmente a rischio di disidratazione ipernatremica, in quanto a questa età può essere inadeguato il riconoscimento della poliuria, della sete e conseguentemente l’apporto idrico. Il rilievo di elettroliti e osmolarità su campioni plasmatici e urinari prelevati contemporaneamente nelle prime ore del mattino orienta la diagnosi: è suggestiva per diabete insipido centrale un’osmolarità urinaria ≤ 300 mOsm/L, associata a osmolarità plasmatica ≥ 300 mOsm/L, soprattutto se in presenza di sodiemia ≥ 145 mEq/L. Il test dell’assetamento è pericoloso e deve essere fatto solo in centri specializzati con il paziente ricoverato.
Risonanza magnetica
La RM dell’encefalo e della zona ipotalamo-ipofisaria senza e con mezzo di contrasto è essenziale nell’iter diagnostico dei neonati con ipopituitarismo sospetto o già diagnosticato, in quanto esiste una correlazione tra anomalie neuroradiologiche e gravità ed evoluzione delle endocrinopatie. I segni da ricercare comprendono: dimensioni dell’adeno-ipofisi, presenza e posizione della neuro-ipofisi (assente o ectopica/non discesa), presenza e morfologia del peduncolo ipofisario, del corpo calloso e del setto pellucido, aspetto dei nervi ottici e del chiasma, anomalie associate (oloprosencefalia, schizencefalia, ipoplasia cerebellare, aplasia del fornice, malformazione di Chiari). Il rischio di ipopituitarismo è 27.2 volte maggiore nei pazienti con neuro-ipofisi ectopica rispetto a quelli in cui è normalmente posizionata.
LA GESTIONE DEL PAZIENTE CON IPOPITUITARISMO CONGENITO
Nelle forme genetiche l’analisi molecolare consente di prevedere quali deficit si possono sviluppare e di ottimizzare i tempi di inizio delle varie terapie sostitutive (le mutazioni di PROP1 comportano la progressiva comparsa di deficit di tutte le tropine ipofisarie, mentre quelle di Pit-1 risparmiano ACTH e gonadotropine).
Il neonato con ipopituitarismo congenito può presentarsi con ipoglicemia persistente e/o micropene e/o ittero colestatico e ipertransaminasemia. La terapia con GH, idrocortisone e levotiroxina normalizza la glicemia e l’ittero colestatico; il trattamento con testosterone enantato, alla dose di 25 mg/mese per 3-4 mesi incrementa le dimensioni dell’asta.
Terapia con glucocorticoidi
L’iposurrenalismo centrale può essere già presente alla nascita o instaurarsi gradualmente, come nell’ipopituitarismo congenito da mutazione di PROP-1. La terapia si basa sulla somministrazione di idrocortisone, analogamente a quella dell’iposurrenalismo primitivo. Si differenzia tuttavia da quest’ultimo per alcuni aspetti fondamentali: non è necessario somministrare un mineralcorticoide, in quanto il sistema renina-angiotensina-aldosterone funziona correttamente. La dose sostitutiva è meno chiaramente individuabile, sia per l’inutilità del dosaggio dell’ACTH nel monitoraggio della terapia, sia per le interferenze reciproche legate al trattamento dei deficit associati. Recentemente, la produzione quotidiana del cortisolo è stata valutata tra 6 e 8 mg/m²/die, pertanto la terapia consigliata è stata ridotta a questo dosaggio, in assenza di episodi ipoglicemici o altri segni di iposurrenalismo. Particolare cautela nello stabilire le dosi deve essere posta con i bambini sotto i 3 anni, per il maggiore rischio di ipoglicemia in caso di supplementazione insufficiente. La somministrazione in 3 dosi quotidiane sembra mantenere livelli di cortisolemia più fisiologici nelle 24 ore. È inoltre necessario utilizzare la minor dose possibile, per evitare un effetto negativo sull’accrescimento staturale.
Sebbene l’iposurrenalismo centrale sia solitamente meno severo di quello primitivo, questi pazienti possono essere a rischio di iposurrenalismo acuto e richiedere adattamento della dose in corso di stress e una preparazione adeguata agli interventi chirurgici.
Terapia con L-tiroxina
La levotiroxina deve essere iniziata non appena posta la diagnosi, alle dosi comunemente utilizzate nell’ipotiroidismo primario, da adeguare sulla base del valore dell’FT4, che deve essere mantenuto nel terzo superiore del range di normalità. Poiché la tiroxina può evidenziare un iposurrenalismo latente, nel caso di un’insufficienza di ACTH è importante iniziare prima la terapia con cortisolo e, nei casi dubbi, seguire il paziente con stretto monitoraggio.
Terapia con GH
Quando si inizia, è necessario monitorare, inizialmente ogni 3 mesi poi ogni 6 mesi, FT3, FT4 e TSH, perché IGHD può mascherare un ipotiroidismo centrale, che si evidenzia dopo l’inizio della terapia sostitutiva con GH. La terapia con GH causa talvolta un aumentato fabbisogno di L-tiroxina in caso di ipotiroidismo centrale già in trattamento. In caso di sospetto deficit ipofisario multiplo e di FT4 a livelli minimi della norma, può essere indicato iniziare la terapia con L-tiroxina prima di quella con GH. È importante ottimizzare la crescita prima dell’età puberale.
Le modifiche terapeutiche della fase di transizione e nell'età adulta sono analoghe a quelle dell’IGHD, mentre la pausa di terapia e il retesting sono consigliati ma non considerati come assolutamente necessari.
Terapia dell’ipogonadismo centrale
Ha lo scopo di indurre la pubertà, mantenere i caratteri sessuali secondari, consentire lo spurt puberale e una regolare mineralizzazione ossea, creare le premesse per un’eventuale fertilità in età adulta.
Nel maschio, l’induzione puberale si inizia a 13-14 anni con 3 possibili schemi terapeutici:
- testosterone enantato im ogni 15 giorni a dosi progressivamente crescenti da 25 a 200-250 mg;
- gonadotropine im (hCG 1-2 volte/settimana e hMG o FSH ricombinante 2-3 volte/settimana);
- gonadotropine per 1-2 anni e successivamente testosterone.
Il testosterone ha maggiore efficacia e rapidità di risultati e migliore compliance, ma comporta mancato aumento del volume testicolare. L’uso combinato dei due schemi in successione è probabilmente il migliore, ma rimane prioritario valutare insieme al ragazzo e ai familiari quale sia la scelta più opportuna. Recentemente la terapia con gonadotropine è stata proposta anche nel periodo neonatale, in alternativa alla terapia con testosterone, per mimare quanto fisiologicamente succede nella “mini-pubertà”.
Nella femmina l’induzione della pubertà avviene verso gli 11-12 anni, con la somministrazione per os a dosi crescenti di etinil-estradiolo (da 2.5 a 15 µg/die) o estrogeni coniugati (da 0.3 a 1.25 mg/die) o di estradiolo per via transdermica (cerotti, gel). Alla comparsa del primo sanguinamento vaginale si passa a somministrare estroprogestinici ciclici.
Diabete insipido centrale
La desmopressina, analogo sintetico dell’ormone antidiuretico, può essere somministrata come spray nasale o per via sublinguale. Il fabbisogno è molto variabile, con dosi di 1-2 spruzzi (5-10 µg) o 1 compressa sublinguale (60 µg) per 1-3 volte al giorno. Il monitoraggio si basa sulla diuresi, che non dovrebbe superare i 1800 mL/m²/die.
Nel neonato e nel piccolo lattante a volte si preferisce soprassedere temporaneamente alla terapia con desmopressina, per la difficoltà di stabilire la dose adeguata e per il rischio di intossicazione da acqua in caso di sovradosaggio. Particolare cautela deve essere posta inoltre nel trattamento del diabete insipido secondario a malformazioni del sistema nervoso centrale, quali l’oloprosencefalia, in cui anche dosi molto basse possono causare ampie variazioni della sodiemia e dell’osmolarità. In tutti questi casi si consiglia in prima istanza di assicurare un apporto idrico sufficiente a compensare le perdite, iniziando poi con dosi di desmopressina estremamente ridotte (es. 1/16- 1/8 di compressa/die) solo in caso di persistente squilibrio idro-elettrolitico.
BIBLIOGRAFIA
- Castinetti F, Reynaud R, Saveanu A, et al. An update in the genetic aetiologies of combined pituitary hormone deficiency. Eur J Endocrinol 2016, 174: R239-47.
- Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I, et al. Hypopituitarism. Lancet 2007, 369: 1461-70.
- Alatzoglou KS, Dattani MT. Genetic forms of hypopituitarism and their manifestation in the neonatal period. Early Hum Dev 2009, 85: 705-12.
- Agha A, Walker D, Perry L, et al. Unmasking of central hypothyroidism following growth hormone replacement in adult hypopituitary patients. Clin Endocrinol 2007, 66: 72-7.
- Maguire AM, Ambler GR, Moore B, et al. Prolonged hypocortisolemia in hydrocortisone replacement regimens in adrenocorticotrophic hormone deficiency. Pediatrics 2007, 120: e164-71.
- Kiess W, Conway G, Ritzen M, et al. Induction of puberty in the hypogonadal girl – practices and attitudes of pediatric endocrinologists in Europe. Horm Res 2002, 57: 66-71.
Ipoparatiroidismi in età pediatrica
Luisa de Sanctis
Dipartimento di Scienze di Sanità Pubblica e Pediatriche, Università di Torino – Ospedale Infantile Regina Margherita- AOU Città della Salute – Torino
La condizione di ipoparatiroidismo è determinata da un deficit di secrezione del paratormone (PTH), ormone chiave nella regolazione del metabolismo calcio-fosforico; è caratterizzata dal punto di vista laboratoristico da ipocalcemia associata a iperfosforemia.
Un tempo tale condizione era di difficile riconoscimento, in quanto i metodi di dosaggio del PTH non permettevano di distinguere l’ormone attivo dai suoi molti metaboliti inattivi circolanti; gli attuali metodi, basati su immunochemiluminescenza, sono invece molto sensibili e specifici, poiché sono in grado di riconoscere la molecola intatta del PTH.
Cause
Diverse sono le cause che portano alla condizione di ipoparatiroidismo (tabella).
| Classificazione degli ipoparatiroidismi |
| Aplasia/ipoplasia delle paratiroidi o anomala produzione di PTH (FIH, del22q11, del10p13, HDR, S. di Kenny-Caffey) Malattie del DNA mitocondriale (S. di Kearns-Sayre, MELAS, MTPDS) Poliendocrinopatia autoimmune tipo I (APS 1, APECED) Lesioni infiltrative (m. di Wilson, Talassemia, Emocromatosi) Chirurgia del collo Deficit di magnesio Alterazione del recettore del Calcio (CaSR, mutazioni attivanti con ipercalciuria) Ipoparatiroidismo idiopatico |
Gli studi di biologia molecolare hanno permesso di individuare i principali difetti genetici responsabili di ridotta sintesi dell’ormone per aplasia/ipoplasia della ghiandola paratiroidea o per difetti genetici nella cascata enzimatica che porta alla formazione dell’ormone.
Alla base dell’ipoparatiroidismo isolato familiare (FIH), disordine classicamente caratterizzato da livelli di PTH ridotti, senza chiari difetti anatomici o disordini anticorpali, sono state descritte forme a carattere autosomico dominante, recessivo e X-linked e sono state identificate mutazioni a carico del gene del PTH (sul cromosoma 11p15.3), del recettore del Calcio (CaSR, cromosoma 3q13.3-q21), di un fattore di trascrizione coinvolto nell’embriogenesi delle paratiroidi (GCM2, cromosoma 6p24.2) e un linkage con il locus HYPX (forma X-linked, Xq26-q27).
Con l’acronimo CATCH22 viene identificato un gruppo eterogeneo di disordini, comprendente la sindrome di Di George e la sindrome Velo-Cardio-Faciale, legato a difetto nella migrazione della cresta neurale cervicale, caratterizzato da ipoplasia delle paratiroidi, ipoplasia del timo, palatoschisi e difetti cardiaci cono-truncali, condizioni associate alla delezione del cromosoma 22q11.2.
Le rare sindromi di Kenny-Caffey e Sanjad-Sakati (che associano all’ipoparatiroidismo la microcefalia) sono legate ad alterazioni del gene TBCE, che codifica per un cofattore importante nel folding della tubulina, mentre la sindrome HDR (con ipoparatiroidismo, deficit uditivo e nefropatia) è legata ad alterazione del gene GATA3, che codifica per un fattore di trascrizione implicato nella via di sintesi del PTH. Sono state recentemente caratterizzate anche le alterazioni mitocondriali alla base delle rarissime sindromi di Kearns Sayre , MELAS e MTPDS.
La produzione di anticorpi anti-paratiroidi determina, invece, il quadro di ipoparatiroidismo autoimmune, che può essere isolato o associato ad altre malattie autoimmuni come il morbo di Addison, la gastrite atrofica e il diabete tipo 1 (Sindrome Polighiandolare Autoimmune tipo 1).
Forme sporadiche di ipoparatiroidismo possono riscontrarsi nel caso di malattie da accumulo di rame (morbo di Wilson) o di ferro (Emocromatosi o Talassemia), per ridotta produzione di PTH da parte delle ghiandole infiltrate.
L’ipoparatiroidismo secondario a danno o asportazione delle paratiroidi durante un intervento chirurgico di tiroidectomia, di paratiroidectomia o altri interventi sul collo per patologie non endocrine, è più raro nell’età evolutiva rispetto a quella adulta, per la differente epidemiologia delle malattie di base.
Un ipoparatiroidismo funzionale, può essere causato da severa ipomagnesemia, in quanto il magnesio è fondamentale per la normale secrezione di PTH. Sono ad oggi peraltro note due forme genetiche di ipoparatiroidismo secondario a ipomagnesemia, una associata a ipercalciuria e nefrocalcinosi (da alterazioni del gene PCLN1, cromosoma 3q27, deputato al trasporto di Mg++/Ca++ a livello tubulare), l’altra a ipocalcemia (causata da alterazioni nel gene TRPM6, 9q22, coinvolto nel trasporto di Mg++ e Ca++ in intestino e reni).
Esiste infine un ipoparatiroidismo idiopatico, definito così perché legato a cause ancora sconosciute.
Clinica
Dal punto di vista clinico l’ipoparatiroidismo si presenta con i segni e sintomi caratteristici dell’ipocalcemia, che, in età pediatrica, sono diversi a seconda se interessino il periodo neonatale e della prima infanzia o le epoche successive.
In età neonatale sono quanto mai aspecifici, essenzialmente legati a una condizione di ipereccitabilità o depressione della conduzione nervosa o della cellula miocardica; spia di ipocalcemia possono essere da un lato l’irritabilità, i tremori fini, l’iper-reflessia, clonie e convulsioni, ma anche scarsa suzione, vomito, crisi di apnea e letargia. Il monitoraggio cardio-respiratorio, a cui spesso sono sottoposti neonati ricoverati nelle terapie intensive, dimostra un allungamento del tratto QT o aritmie nel tracciato ECG; la depressione della conduzione cardiaca aumenta in questi neonati il rischio di morte improvvisa. I segni di Chvostek (contrazione dei muscoli ai margini delle labbra alla percussione del nervo facciale) e di Trousseau (contrazione dei muscoli della mano indotta dalla compressione del braccio con uno sfigmomanometro mantenuto per 3 minuti a una pressione > 20 mmHg rispetto alla sistolica) nell’età neonatale non sono significativi, mentre diventano premonitori di una crisi tetanica (“tetania latente”) nelle epoche successive. In queste i sintomi di ipocalcemia sono tuttavia variabili, da lievi a severi, in base alla gravità e alla rapidità di insorgenza dell’alterazione: da parestesie e formicolii a mani, piedi o intorno alla bocca, contrazioni spastiche con crampi dolorosi a gruppi muscolari diversi, fino a un quadro generalizzato di tetania e spasmo laringeo nelle forme a insorgenza acuta o rapida; pelle secca e spessa, unghie fragili e discromiche, ansia, irritabilità, labilità emotiva, fino a depressione o forme psicotiche nelle forme ad andamento cronico.
Nelle forme croniche nel tempo possono comparire disturbi specifici secondari alla deposizione di fosfato tricalcico nel nuclei centrali della base (tremori e rigidità), del cristallino (cataratta), nel tessuto sottocutaneo e nei tendini (nodosità visibili o palpabili).
Diagnosi
Il laboratorio evidenzia bassi valori di paratormone (PTH) in presenza di calcio basso (ipocalcemia) e fosforo alto (iperfosforemia). L’eliminazione di calcio nelle urine (calciuria) può essere alta se la calcemia è molto bassa.
L'esame radiologico del cranio, la TC o la RM encefalo possono evidenziare la presenza di calcificazioni nei nuclei della base.
All’ECG l'ipocalcemia prolunga l'intervallo QT.
La diagnosi differenziale è con tutte le altre condizioni con ipocalcemia che si caratterizzano, tuttavia, per la presenza di valori elevati di PTH: pseudo-ipoparatiroidismo, insufficienza renale cronica, deficit di vitamina D, tubulopatie con perdita di calcio, malassorbimenti, pancreatite acuta, metastasi osteoblastiche). Esistono, tuttavia, forme familiari, legate all’alterata funzione del recettore del calcio, caratterizzate da ipocalcemia e ipercalciuria con PTH normale-basso, che devono essere distinte in quanto la terapia con il calcio determina un peggioramento della calciuria e della funzione renale.
Terapia
La terapia dell’ipoparatiroidismo mira a correggere l’ipocalcemia, principalmente attraverso la somministrazione di calcio e vitamina D.
In commercio esistono diversi preparati di calcio (gluconato, cloruro, citrato, lattato e carbonato), che trovano diversa indicazione, a seconda dell’età del paziente, dell’entità dell’ipocalcemia e della sintomatologia ad essa legata.
In età neonatale viene perlopiù utilizzato il Ca gluconato al 10%, per via endovenosa o per via orale. La via endovenosa è da preferire nel caso di ipocalcemia severa (calcemia < 7 mg/dL nel nato a termine; < 6 mg/dL nel pretermine) o associata a sintomatologia importante (alterazioni all’ECG, tachicardia, tremori, crisi convulsive). La dose di attacco prevede l’utilizzo di 2 mL/kg in 10-15 minuti, ripetibile dopo 4-6 ore, sotto stretto monitoraggio cardiaco (per il possibile rischio di bradicardia, fino all’arresto cardiaco) ed elettrolitico. È preferibile l’utilizzo di un accesso venoso centrale, in quanto lo stravaso di calcio nel tessuto sottocutaneo può determinare necrosi tissutale, con ulcere cutanee. La terapia dell’ipocalcemia neonatale meno severa o asintomatica e quella di mantenimento prevedono l’utilizzo di Ca Gluconato al 10% per bocca, al dosaggio di 4-5 mL/kg/die (che corrisponde a circa 40 mg/kg/die).
Nelle età successive a quella neonatale il dosaggio del Ca gluconato al 10% nella fase acuta è variabile (5-20 mg/kg di Ca elemento, in 5-15 minuti, ripetibile ogni 4-6 ore); per la terapia cronica domiciliare viene solitamente utilizzato il Calcio carbonato, a un dosaggio variabile a seconda delle forme, della risposta e del momento di crescita, da 50-100 mg/kg o 400-800 mg ogni 6-12 ore. Per aumentare l’assorbimento, ridurre l’oscillazione delle concentrazioni nell’arco della giornata e gli effetti collaterali legati al sovradosaggio di questa terapia cronica, è importante che l’assunzione del Calcio venga frazionata nell’arco della giornata.
La vitamina D è il farmaco più efficace nella terapia dell’ipoparatiroidismo, perché stimola il riassorbimento di calcio dall’intestino e controlla il riassorbimento del tessuto osseo. Nel caso degli ipoparatiroidismi è necessario somministrare il metabolita attivo della vitamina D (calcitriolo), o l’alfa-calcidiolo, in quanto, in assenza di PTH, la vitamina D non viene attivata a livello renale. Per la riduzione dell’attività dell’1-alfa-idrossilasi renale, il dosaggio di calcitriolo richiesto può essere superiore rispetto ad altre condizioni, oscillando tra 20-60 ng/kg/die (0.25-2 µg/die); tuttavia può variare nel tempo e va adattato ai valori della calcemia.
Le forme di ipoparatiroidismo secondarie a ipomagnesemia necessitano la supplementazione di questo ione, sottoforma di magnesio solfato.
Recentemente l’AIFA ha autorizzato in Italia l'uso di paratormone sintetico ricombinante (rhPTH) per il trattamento delle forme più severe e refrattarie di malattia; mimando il meccanismo fisiologico, l’ormone sostitutivo potrebbe rappresentare la soluzione alla cura di questa patologia cronica, riducendo così le temibili complicanze renali legate all’assunzione di vitamina D e Ca a lungo nel tempo. Sono necessari ulteriori studi, a lungo termine e su più ampie casistiche, anche pediatriche, per confermare la superiorità di questo trattamento rispetto alle terapie attuali.
Follow-up
Il monitoraggio della terapia deve essere biochimico 2-3 volte/anno (più frequente in caso di modifica della terapia):
- calcemia: deve essere mantenuta sui livelli bassi della norma (8.5-8.8 mg/dL);
- fosfatemia: nel caso di livelli > 6 mg/dL è necessaria la chelazione con anti-acidi a base di idrossido di alluminio, per prevenire la deposizione di calcificazioni eterotopiche;
- creatininemia (per valutare la funzionalità renale);
- calciuria delle 24h: deve essere < 4 mg/kg/die;
- rapporto calciuria/creatininuria: deve essere mantenuto < 0.21.
Deve essere programmata ecografia renale (per controllare l’assenza di nefrocalcinosi) 1-2 volte l’anno, più sovente in caso di ipercalciuria o aumentato rapporto calciuria/creatininuria.
Bibliografia
- Al-Azem H, Khan AA. Hypoparathyroidism. Best Pract Res Clin Endocrinol Metab 2012, 26: 517-22.
Displasie scheletriche
Tiziana Greggi
Chirurgia delle Deformità del Rachide, Istituto Ortopedico Rizzoli, Bologna
(aggiornato al novembre 2022)
INTRODUZIONE
Le displasie scheletriche sono un vasto (oltre 400 tipi) ed eterogeneo gruppo di anomalie delle modalità di crescita e sviluppo del tessuto osteo-cartilagineo delle ossa, da causa generalmente genetica. Sono note da numerosi decenni, soprattutto in ambiente ortopedico-pediatrico per la necessità di trattamenti chirurgici.
L’espressività clinica varia dalle forme letali in epoca peri-natale, a bassa statura, talvolta disarmonica, a condizioni di lieve ritardo di crescita. La caratterizzazione clinico-radiologica risulta spesso semplice e ben schematizzabile, mentre la definizione pato-molecolare è molto complessa.
La prevalenza varia molto nei diversi studi (da 2.1 a 4.7/10mila nati), ma quella ritenuta più reale è di 2 casi su 10mila nati vivi e 20 casi su 10mila nati morti (1). Le forme più comuni sono l’acondroplasia, l'acondrogenesi, la displasia tanatofora e l’osteogenesi imperfetta.
Per gli aspetti anatomici, istologici e di struttura del tessuto osseo bisogna fare un breve cenno ai due processi di sviluppo del tessuto osseo e ad alcune fasi dell'embriogenesi. Circa alla 5° settimana di gestazione compaiono gli arti superiori e, qualche giorno dopo, gli arti inferiori; alla 7° appare la cartilagine, alla 12° sono presenti i centri di sviluppo delle ossa lunghe (fig 1).
Figura 1
Sviluppo del feto: A = 5.5 settimane; B = 7 settimane; C =12 settimane
L’osso dello scheletro si genera per:
- ossificazione diretta membranosa del mesenchima, a formare le ossa della volta cranica e delle clavicole, in cui le cellule progenitrici mesenchimali si differenziano via pre-osteoblasti in osteoblasti;
- ossificazione indiretta encondrale (cartilaginea) delle ossa lunghe, vertebre e coste, dove le cellule mesenchimali progenitrici si differenziano dapprima in cellule pericondriali e condrociti e successivamente si trasformano in osteoblasti.
Per chiarire lo schema di ossificazione encondrale: alle estremità delle ossa lunghe c’è la cartilagine di accrescimento, dove si distinguono 4 zone cellulari (di riserva, di proliferazione, pre-ipertrofica e ipertrofica), in cui si completa il modello cartilagineo che subirà l’apposizione di tessuto osseo e la sua sostituzione con l’invasione contemporanea dei vasi sanguigni. La crescita in senso latero-laterale è dovuta alle cellule pericondriali, che diverranno cellule del periostio nell’osso maturo (fig 2).
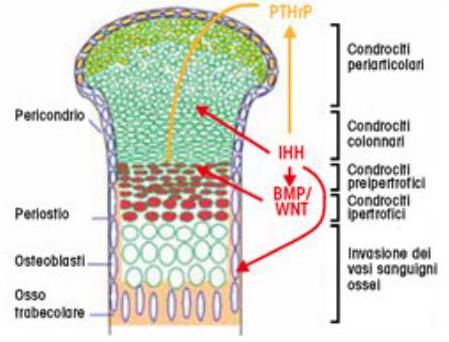
Figura 2
Via di segnale che determina la crescita della cartilagine di accrescimento e la formazione del tessuto osseo. Lo sviluppo della cartilagine di accrescimento è determinato dal controllo di diverse vie di segnale, tra cui IIHH e PTHrP, BMPs, WNT. Tutte inducono lo sviluppo della zona di proliferazione dei condrociti e ne inibiscono la maturazione, contrastando l'azione contraria della via FGFs/FGFr. Lo stampo cartilagineo verrà poi riassorbito dall'azione di metallo-proteasi, sostituito da osteoblasti e vasi sanguigni, quindi da tessuto osseo maturo.
È importante ricordare che il tessuto osseo è dinamico: grazie all’equilibrio funzionale delle due linee cellulari (osteoblasti e osteoclasti), subisce un continuo e regolare processo di riassorbimento e neoformazione (2).
Cercherò qui di presentare sinteticamente questo vastissimo argomento, puntualizzando le principali caratteristiche di queste condizioni patologiche, facendo riferimento alle forme più comuni e alle classificazioni più attuali.
CLASSIFICAZIONE
Ci sono stati numerosi tentativi di portare ordine in questo campo, elaborando una classificazione delle malattie costituzionali dello scheletro, contenente al primo posto il capitolo delle osteo-condro-displasie (3,4). Secondo una recente revisione del 2019 del gruppo di esperti dell’International Skeletal Dysplasias Society, sono classificate 461 condizioni di displasia scheletrica, suddivise in 42 diversi gruppi con specifici criteri clinici, radiografici e/o molecolari (5) (tab 1).
| Tabella 1 Classificazione delle displasie scheletriche genetiche (5) |
||
| # | Gruppo | Esempi |
| 1 | Condro-displasie correlate a FGF-R3 | Acondroplasia, ipocondroplasia |
| 2 | Collagene di tipo 2 | S. di Stickler |
| 3 | Collagene di tipo 11 | Fibrocondrogenesia |
| 4 | Disordini della solfatazione | S. di Ehlers-Danlos |
| 5 | Perlecan | Condrodistrofia miotonica |
| 6 | Aggrecan | Ipostaturismo con età ossea avanzata |
| 7 | Filamina | Displasia fronto-metafisaria |
| 8 | TRPV4 | Displasia metatropica |
| 9 | Ciliopatie | Displasia toracica asfissiante |
| 10 | Displasie epifisarie multiple e pseudo-acondroplasia | Pseudo-acondroplasia |
| 11 | Displasie metafisarie | S. di Shwachman–Bodian–Diamond |
| 12 | Displasie spondilo-metafisarie | Spondilo-encondrodisplasia |
| 13 | Displasie spondilo-epi-(meta)-fisarie | Displasia di Dyggve–Melchior–Clausen |
| 14 | Displasie spondilo-displasiche severe | Acondrogenesia |
| 15 | Displasie acromeliche | Acrodisostosi |
| 16 | Displasie acro-mesomeliche | Displasia acro-mesomeliche di Maroteaux |
| 17 | Displasie mesomeliche e rizo-mesomeliche | Discondroplasia di Leri-Weill |
| 18 | Displasie con incurvamento osseo | Displasia camptomelica |
| 19 | Nanismi primitivi con ossa sottili | S. di Kenny-Caffey, s. IMAGE |
| 20 | Displasie con dislocazioni articolari multiple | Displasia di Desbuquois |
| 21 | Condrodisplasia puntata | S. di Keutel |
| 22 | Displasie osteosclerotiche neonatali | Displasia di Blomstrand |
| 23 | Osteopetrosi | Osteopetrosi neonatale severa, picnodisostosi |
| 24 | Disordini osteosclerosanti | Displasia diafisaria di Camurati-Engelman |
| 25 | Osteogenesi imperfetta | Osteogenesi imperfetta tipi 1-6 |
| 26 | Anomalie della mineralizzazione | Ipofosfatasia, rachitismi ipofosfatemici |
| 27 | Malattie da accumulo lisosomiale con coinvolgimento osseo | Mucopolisaccaridosi |
| 28 | Malattie osteolitiche | Progeria di Hutchinson-Gilford |
| 29 | Sviluppo disorganizzato delle componenti scheletriche | S. di McCune-Albright, neurofibromatosi tipo 1, fibrodisplasia ossificante progressiva |
| 30 | Alta statura con coinvolgimento scheletrico | S. di Sotos, s. di Marfan |
| 31 | Osteoartropatie genetiche infiammatorie simil-reumatiche | Displasia pseudo-reumatoide progressiva |
| 32 | Displasie cleido-craniche | Displasia cleido-cranica |
| 33 | Cranio-sinostosi | S. di Crouzon |
| 34 | Disostosi con coinvolgimento cranio-facciale predominante | S. di Treacher-Collins |
| 35 | Disostosi con predominante coinvolgimento vertebrale (con e senza coinvolgimento costale) | S. di Klippel-Feil |
| 36 | Disostosi rotulee | S. della rotula piccola |
| 37 | Brachidattilie (senza manifestazioni extra-scheletriche) | S. di Cooks |
| 38 | Brachidattilie (con manifestazioni extra-scheletriche) | Pseudoipoparatiroidismo tipo IA |
| 39 | Ipoplasie degli arti | Sirenomelia |
| 40 | Ectrodattilia | S. di Hartsfield |
| 41 | Polidattilia-sindattilia-trifalangismo | S. di Pallister-Hall |
| 42 | Difetti della formazione articolare e sinostosi | S. di Liebenberg |
CONSIDERAZIONI GENERALI CLINICO-DIAGNOSTICHE E DI TRATTAMENTO
Ciascun tipo di displasia scheletrica è caratterizzato da un insieme diverso di anomalie.
Gli aspetti clinici comuni fondamentalmente sono: bassa statura, braccia e gambe corte, dita corte, testa sproporzionatamente grande, collo corto, mobilità ridotta all’altezza dei gomiti. Alcuni tipi di displasia scheletrica vengono rilevati a circa 20 settimane di gravidanza nel corso di un’ecografia, mentre altri possono non risultare evidenti fino alla prima infanzia. Anche se la displasia scheletrica viene rilevata nel corso della gravidanza, può risultare difficile diagnosticarne il tipo esatto prima del parto, soprattutto per la difficoltà di approfondimenti diagnostico-strumentali, meglio eseguibili quando i problemi della crescita si presentano in età pediatrica o nella prima infanzia (6,7).
Per quel che riguarda la prognosi, non c'è sopravvivenza quando la displasia scheletrica causa crescita ossea particolarmente anomala, che impedisce al torace e ai polmoni di svilupparsi in modo corretto. Tale condizione caratterizza le displasie scheletriche letali e si verifica in circa 1 neonato su 10mila. Si definisce invece non letale la displasia scheletrica in cui i neonati colpiti sopravvivono al parto e al periodo immediatamente successivo. Tra i bambini affetti da displasia scheletrica non letale, le diagnosi più comuni sono l’acondroplasia, spesso indicata con il nome di nanismo, e l'osteogenesi imperfetta detta anche "malattia delle sclere blu".
Il sospetto diagnostico viene posto inizialmente tramite la raccolta dell’anamnesi familiare e personale del bambino e l’esame obiettivo, con particolare attenzione alla valutazione auxologica e alla ricerca di alcuni dismorfismi caratteristici. In particolare, la valutazione clinica deve comprendere la rilevazione dei seguenti parametri: peso, lunghezza e circonferenza cranica alla nascita, valutazione della curva di crescita, sia staturale sia ponderale, e della curva di aumento della circonferenza cranica, rapporto tra segmento corporeo superiore e inferiore, descrizione dell’eventuale accorciamento delle estremità, caratteristiche del cranio, anomalie dentali, scoliosi, lordosi, varismo o valgismo del ginocchio. Dovrebbero essere valutate inoltre le caratteristiche di pelle, unghie e capelli, insieme a disturbi cognitivi e potenziali anomalie dell’udito e/o della vista.
La conferma diagnostica si ottiene tramite test genetici specifici. Dovrebbero essere eseguiti esami ematici generali, oltre a test renali, epatici e cardiaci per valutare l’eventuale presenza di complicanze a carico di tali organi. È necessaria una valutazione radiologica per definire la gravità della patologia e l’eventuale presenza di complicanze (8,9).
Il trattamento delle displasie scheletriche è multi-disciplinare. La gestione del bambino può essere ottimizzata utilizzando un team specialistico che dovrebbe coinvolgere pediatri, genetisti, neurologi e neurochirurghi pediatrici, pneumologi pediatrici e chirurghi ortopedici pediatrici.
L’assistenza preventiva è essenziale e ogni sforzo deve essere diretto a identificare i bambini ad alto rischio di sviluppare complicanze, in modo da mettere in atto interventi terapeutici appropriati e precoci, per prevenire sequele permanenti.
Negli ultimi anni si sono delineate nuove promettenti prospettive terapeutiche, mirate ai meccanismi alla base della malattia e non solo alla correzione chirurgica dei difetti ad essa associati. L’intervento chirurgico è una forma comune di terapia per l’ipostaturismo (nanismo), sia armonico sia disarmonico. Tuttavia, l’indicazione chirurgica rimane controversa, poiché si tratta di una procedura dolorosa e associata a complicanze, che includono infezioni, contratture muscolari e aumento del rischio di fratture (10,11). L’allungamento dell’arto attraverso la procedura di Ilizarov comporta, infatti, la rottura chirurgica delle ossa lunghe, la fissazione esterna e la distrazione graduale per molti mesi durante il processo di guarigione. La lunghezza media ottenuta è di circa 20.5 cm dopo multiple procedure applicate sia al femore sia alla tibia. È necessaria un’attenta valutazione clinica e psicologica pre-operatoria, per valutare l’alto rischio di complicanze rispetto al miglioramento staturale ottenibile. Le recenti innovazioni, come l’uso della fissazione intra-midollare, possono migliorare i risultati e ridurre i rischi (12-14). In futuro, la combinazione dell’allungamento chirurgico dell’arto con le strategie farmacologiche potrebbe condurre a un ulteriore miglioramento della prognosi staturale di questi pazienti.
Attualmente non esistono terapie farmacologiche approvate per le condro-displasie, ad eccezione dell’ormone della crescita, che ha indicazione per l’acondroplasia soltanto in Giappone. Negli ultimi anni sono state proposte e studiate molte strategie non chirurgiche, sfruttando l'impiego di anticorpi monoclonali volti a ridurre l’eccessiva attivazione di FGF-R3, in modo da stimolare la crescita ossea lineare nei pazienti affetti da acondrodisplasia, o di recente la disponibilità di un anticorpo monoclonale, il burosumab, per i pazienti con la forma di rachitismo ipofosfatemico X-linked (11,15,16). Nonostante ci troviamo già in fase di sperimentazione di numerose nuove promettenti terapie farmacologiche (11,16-18), oggi per cercare di migliorare la qualità e la durata della vita di chi è affetto da displasia scheletrica bisogna spesso considerare l'impiego di soluzioni chirurgiche. È tuttora attuale la necessità di correggere chirurgicamente la direzione in cui crescono le ossa o per allungare gli arti, per eseguire osteo-sintesi di fratture ripetute, per correggere la colonna vertebrale, per allargare il canale vertebrale, per inserire uno shunt atto a drenare l’ipertensione endo-cranica.
I trattamenti riabilitativi fisiatrici sono di estrema necessità per rafforzare i muscoli e aumentare l’estensione dei movimenti. Per il trattamento di questi stati patologici è indispensabile la messa a punto e personalizzazione di attrezzature di assistenza e modi alternativi per lo svolgimento delle attività quotidiane. I chirurghi ortopedici pediatrici, i neurochirurghi e i fisiatri non sono gli unici operatori, ma agiscono in collaborazione con genetisti, cardiologi, otorinolaringoiatri, oftalmologi, neurologi, neuropsichiatri infantili, endocrinologi, fisiatri, avvalendosi inoltre di un adeguato counseling e supporto psicologico.
SISTEMATICA DELLE DISPLASIE SCHELETRICHE: EZIOLOGIA E DIAGNOSI
Queste patologie possono derivare da difetti genetici, noxae esogene o da sindrome della banda amniotica.
Evitiamo di affrontare la classificazione molecolare molto estesa e accurata, basata sulle diverse alterazioni genetiche presenti nelle displasie scheletriche, che agiscono sulla cartilagine di accrescimento, limitandoci a segnalare che possono essere anomalie genetiche che agiscono nello sviluppo della cartilagine di accrescimento o coinvolgono la sintesi di proteine extra-cellulari (tab 2-4).
| Tabella 2 Anomalie genetiche che agiscono nello sviluppo della cartilagine di accrescimento |
|||
| Condizione | Geni | Trasmissione | Segni clinici e radiologici |
| Segnale FGF | |||
| Acondroplasia | FGF-R3 | Autosomica dominante (AD) | Macrocrania, ipoplasia media facies, bozze frontali prominenti. Riduzione rizomelica (parte prossimale) degli arti, limitata estensione del gomito, mano a tridente, ginocchio varo, iperlordosi lombare. Ossa lunghe corte e ricurve, pelvi a tridente, ridotta distanza fra i peduncoli vertebrali lombari. |
| Displasia tanatofora tipo I e II | FGF-R3 | AD | Forma letale nel periodo peri-natale, arti molto corti, femori a cornetta del telefono, marcata plati-spondilia e ristrettezza del torace, cranio a trifoglio (tipo II). |
| Ipocondroplasia | FGF-R3 | AD | Ritardo di crescita pre- e post-natale, intelligenza nella norma. Ipoplasia rotulea, iperlassità articolare. Enfisema, tracheo-laringo-bronco-malacia. Difficoltà respiratorie e nell'alimentazione nel primo anno di vita. Anomalie genitali, ipoplasia mammaria. |
| Segnale BMP | |||
| Brachidattilia A1, A2, C | IHH, GDF5, BMPR1B, BMP2, CDMP1 | AD | Anomalie di tutte le falangi medie e fusione con le distali (A1). Anomalie della falange media dell'indice e del secondo dito del piede (A2). Anomalie falangi medie II e III dito mano, anomalie solo falange media V dito, II e IV dito più lungo. |
| Segnale WNT | |||
| Sindrome di Robinow | WNT5A DWL1 DWL3 |
AD RS1 AD RS2 AD RS3 |
Esordio post-natale, bassa statura, brevità acro-mesomelica degli arti, macrocefalia, bozze frontali, occhi prominenti, ipertelorismo, narici antiverse. Appiattimento della regione media della facies. Anomalie dentali (malocclusione, ipodonzia, ritardo eruzione denti permanenti, denti sovrannumerari).
|
| ROR2 | Autosomica recessiva (AR) | Possibili anomalie cardiache e renali, labio-palatoschisi, displasie ungueali. Riduzione mesomelica soprattutto degli arti superiori, brachidattilia. Segmentazione vertebrale. Una variante allelica AD ha brachidattilia con falangi medie corte, falangi distali rudimentali o assenti, deformità del pollice, alluci grandi. |
|
| Segnale PTHrO | |||
| Displasia acro-capito-femorale | IHH | AR | Bassa statura disarmonica, macrocrania relativa, brachidattilia, epifisi a cono alle mani e femore. La forma allelica AD (causata non solo da IHH, ma anche da GDF5 e BMPR1B) presenta brachidattilia tipo A1, tipica per le anomalie di tutte le falangi medie fuse con le distali. |
| Displasia metafisaria tipo Jansen | PTH-R1 | AD | Bassa statura disarmonica grave, faccia prominente con mandibola piccola, arti corti e curvi. Ipercalcemia e iposfatemia. |
| Condrodisplasia tipo Blornstrand | PTH-R1 | AD | Displasia letale, polidramnios, idrope fetale. Anomalie facciali, arti molto corti. Incremento della densità ossea, con maturazione scheletrica avanzata. |
| Segnale CNP/NPR2 | |||
|
Displasia acromesomelica tipo Maroteaux |
NPR2 | AR | Bassa statura disarmonica, con riduzione dei segmenti mesomelici ed acromelici dei quattro arti. Fronte prominente, con naso piccolo e largo. Pectus escavatum/carenato, ipercifosi dorsale, iperlordosi lombare. Intelligenza normale. |
| Bassa statura idiopatica armonica | NPR2 | AD | Bassa statura moderata. Riduzione segmenti mesomelici arti superiori ed inferiori. |
| Tabella 3 Mutazioni genetiche con ruolo non ancora ben definito nella replicazione del DNA |
|||
| Condizione | Geni | Trasmissione | Segni clinici e radiologici |
| Sindrome di Kenny-Caffey | FAM11A | AD | Importante bassa statura, anomalie facciali e oculari, mani e piedi piccoli. Ispessimento della corticale delle ossa lunghe. Stenosi midollare, cranio-stenosi, ritardo chiusura fontanella anteriore. Ipoparatiroidismo, ipocalcemia, possibile ipofosfatemia e anemia. Calcificazioni renali e dei nuclei della base. Possibile epilessia. Intelligenza normale, voce con timbro acuto. |
| Discondrosteosi (sindrome di Léri-Weill) | SH0X | Aplo-insufficienza | Bassa statura mesomelica (brevità degli arti nella porzione media), anomalia di Madelung (curvatura dell'avambraccio per disallineamento fra radio, ulna e ossa carpali). Intelligenza nella norma. |
| Displasia mesomelica di Langer | SH0X | Aplo-insufficienza | Bassa statura severa, brevità delle ossa lunghe. Ipoplasia ulna e perone. Anomalia di Madelung assente. Intelligenza normale. |
| Tabella 4 Difetti delle proteine della matrice extra-cellulare |
|||
| Condizione | Geni | Trasmissione | Segni clinici e radiologici |
| Aggrecano | |||
| Displasia spondilo-epi-metafisaria tipo aggrecano | ACAN | AR | Bassa statura armonica nell'infanzia, disarmonica nell'età adulta. Macrocefalia relativa. Marcata ipoplasia della regione centrale della faccia, con assenza della cartilagine nasale, prognatismo. Collo corto. Torace a botte. Platispondilia e schisi dei corpi vertebrali cervicali. Scoliosi e iperlordosi lombare. Riduzione rizo-mesomelica degli arti, con epifisi irregolari e metafisi allargate. Brachidattilia mani, con pollici larghi. |
| Displasia spondilo-epifisaria tipo Kimberley | ACAN | AD | Habitus tarchiato, con bassa statura armonica. Irregolarità delle cartilagini di accrescimento. Sclerosi dei corpi vertebrali e anomalie epifisarie con ritardo dell'età ossea. Artropatia progressiva severa e precoce. |
| Osteocondrite dissecante familiare | ACAN | AD | Bassa statura disarmonica. Riduzione degli spazi inter-vertebrali. Frammentazione della cartilagine articolare e dell'osso subcondrale, con frammenti liberi intra-articolari, versamento articolare con articolazione a scatto. |
| Bassa statura con età ossea avanzata | ACAN | AD | Età ossea avanzata con blocco anticipato della crescita staturale. |
| Collagene tipo 1 | |||
| Osteogenesi imperfetta | COL1A1 | AD |
Tipo I lieve: fragilità ossea, sclere blu, ipoacusia. Raramente bassa statura e dentinogenesi imperfetta. |
| IFITM5 | AD | Tipo V moderata: fragilità ossea, iperplasia del callo osseo, calcificazione della membrana inter-ossea, assenza di sclere blu e dentinogenesi imperfetta. | |
| Collagene tipo 2 | |||
| Displasia spondilo-epifisaria congenita | COL2A1 | AD |
Bassa statura disarmonica, con prevalenza di tronco corto. Facies piatta, ponte nasale sottile, talora palatoschisi, miopia. Collo corto, petto carenato, ginocchio valgo. Quadro radiologico diverso in rapporto all'età:
|
| Displasia di Kneist | COL2A1 | AD | Faccia mediana piatta, radice nasale infossata, palatoschisi, miopia. Tronco corto, iperlordosi lombare, scoliosi. Arti corti con articolazioni prominenti. Segni radiologici specifici: plati-spondilia, corpi vertebrali con cuneizzazione anteriore, "cleft" coronale lombare. Anomalie epifisarie ossa lunghe, collo femorale corto e largo. |
| Collagene tipo 9 | |||
| Displasia epifisaria multipla |
COL9A1 |
AD |
Bassa statura disarmonica con arti corti. Dolore e tumefazione articolare. Osteo-artrite a esordio precoce. Deambulazione dondolante (anserina). Coinvolgimento di tutte le epifisi, in particolare anca, ginocchio, caviglia, mani e polsi. Appiattimento delle epifisi con l'età, metafisi normali, lieve riduzione di lunghezza ossa lunghe, vertebre con minime anomalie. Segno particolare: rotula bipartita. |
| Pseudo-acondroplasia | COMP | AD | Bassa statura disarmonica. ossa lunghe incurvate. Brachidattilia, con iperlassità soprattutto inter-falangea e carpale. Anomalie vertebrali. Osteo-artrite. Assenza delle caratteristiche facciali proprie dell'acondroplasia. Segni radiologici tipici sono i corpi vertebrali biconvessi, con protrusione anteriore linguiforme, corpi vertebrali normali in adolescenza, anomalie epifisarie diffuse, soprattutto del femore prossimale. |
| Sindrome di Stickler | tipo 1 COL2A1 tipo 2 COL1A1 tipo 3 COL1A2 |
AD |
Displasia ossea spondilo-epimetafisaria. Iperlassità articolare. Osteo-artrite progressiva. Micro-retrognatia, con anomalie del palato, sequenza di Pierre Robin. Ipoacusia. Miopia grave, con distacco di retina ed anomalie del corpo vitreo. |
| COL9A1 COL9A2 COL9A3 |
AR | ||
DIAGNOSI PRE-NATALE
Sono fondamentali: l'anamnesi familiare ed ostetrica, con la datazione corretta della gestazione, unite alla misurazione delle ossa lunghe e delle dimensioni del cranio (circonferenza cranica — HC, circonferenza toracica — CC, circonferenza addominale — AC, lunghezza femorale — FL).
L'esame ecografico nelle diverse fasi della gravidanza permette di:
- I trimestre: identificare gli arti e i diversi segmenti ossei;
- II trimestre: valutare lunghezza, morfologia, motilità, atteggiamento ed ecogenicità dei quattro arti;
- III trimestre: valutare lunghezza degli arti e mineralizzazione ossea.
Nel sospetto di ritardo di crescita intra-uterino (IUGR), misurare il rapporto FL/piede (fig 3).
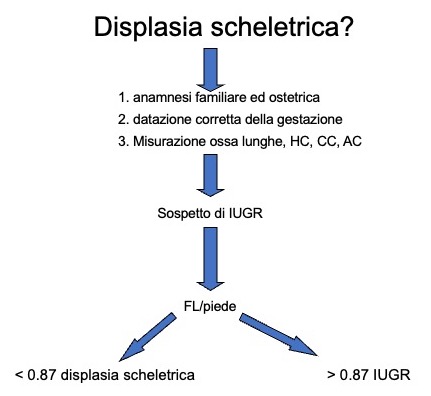
Figura 3
Iter diagnostico per differenziare displasie scheletriche da IUGR
La bassa statura è uno degli elementi clinici per cui più frequentemente un paziente viene sottoposto all’attenzione del pediatra. La Società Europea di Endocrinologia Pediatrica (ESPE) ha proposto nel 2007 una classificazione della bassa statura, aggiornata nel 2016, che la suddivide in tre grandi categorie (5):
- primaria, che comprende le condizioni sindromiche su base genetica;
- secondaria, che comprende i nati piccoli per età gestazionale, con deficit di recupero della crescita;
- idiopatica, che comprende le displasie scheletriche.
Le displasie scheletriche si differenziano come gravità fra:
- forme letali, che si caratterizzano per FL/AC < 0.16, polidramnios, micromelia severa, ipoplasia toracica severa;
- forme non letali, in cui è comunque fondamentale la comparazione con gli aspetti standard dei segmenti ossei, la valutazione qualitativa dell’osso (demineralizzazione, fratture), la valutazione di mani, piedi e cranio (macrocrania, prominenza delle bozze frontali, ipo-ipertelorismo), liquido amniotico, colonna vertebrale, visceri, ecocardiogramma, movimenti fetali.
Il test molecolare e il counseling genetico completano la diagnosi.
DISPLASIE SCHELETRICHE LETALI (0.95-1/10mila nati)
Acondrogenesi
Genetica: deriva da una rara mutazione del gene codificante per il collagene tipo II. Ne esistono due sottotipi, il tipo I (AR) e il tipo II (AD).
Clinica: micromelia estrema.
Diagnosi: ossa appena evidenziabili e ricurve, ipoplasia toracica severa, ipomineralizzazione di grado variabile.
Esito: letale nel 100% dei casi.
Displasia tanatofora
Genetica: deriva da una mutazione relativamente frequente del gene codificante per Fibroblast Growth Factor Receptor.
Clinica: rizomelia severa, ipoplasia toracica, cranio a trifoglio (in una delle varianti).
Esito: letale nel 100% dei casi per ipoplasia toracica.
Osteogenesi imperfetta
Comprende un gruppo eterogeneo di patologie congenite (tab 5), caratterizzate da estrema fragilità ossea, per mutazione dei geni codificanti per il collagene tipo I.
Dopo le prime descrizioni del 1788, sono stati introdotti vari sinonimi: malattia di Lobstein, malattia di Vrolik, osteosatirosi, fragilitas ossium.
| Tabella 5 Classificazione clinica e radiografica dell’osteogenesi imperfetta |
|||
| Tipo | Trasmissione | Statura | Gravità e manifestazioni |
| I | AD | Normale | Lieve: fragilità ossea, sclere blu, ipoacusia (raramente bassa statura e dentinogenesi imperfetta). |
| II | AD | Normale | Letale nel periodo peri-natale, sclere blu, assenza di mineralizzazione, ossa curve con fratture pre-natali, insufficienza respiratoria. |
| III | AD | Bassa | Severa con deformità: scoliosi, spondilo-listesi, schiacciamenti vertebrali, instabilità cervicale C1-C2, invaginazione del dente dell’epistrofeo nella base cranica, con compressione del ponte e manifestazioni neurologiche anche severe (atassia, disfagia), sclere blu, dentinogenesi imperfetta. |
| IV | AD | Bassa | Da moderata a severa fragilità ossea, con fratture frequenti, ma meno numerose alle ossa lunghe, non sclere blu. |
| V | AD | Lievemente diminuita | Fragilità ossea da lieve a moderata, formazione di callo ipertrofico dopo frattura e calcificazione della membrana interossea dell’avambraccio, dentinogenesi imperfetta, assenza di sclere blu. |
| VI | AD | Normale o lievemente diminuita | Alta fragilità ossea con fratture frequenti sin dall’infanzia, fratture vertebrali da compressione, deformità degli arti. Assenza di sclere blu e difetti della dentinogenesi. |
| VII | AR | Variabile | Fratture intra-uterine, deformità degli arti inferiori, accorciamento rizomelico degli arti inferiori, coxa vara. |
Le caratteristiche principali sono 4: difetto genetico, collagenopatia, fragilità ossea, fratture frequenti. Oltre all'osso, sono colpiti altri tessuti, di cui il collagene I è il principale costituente: sclere, dentina, ossa dell’apparato acustico, cute, vasi, capillari, valvole cardiache (fig 4,5).
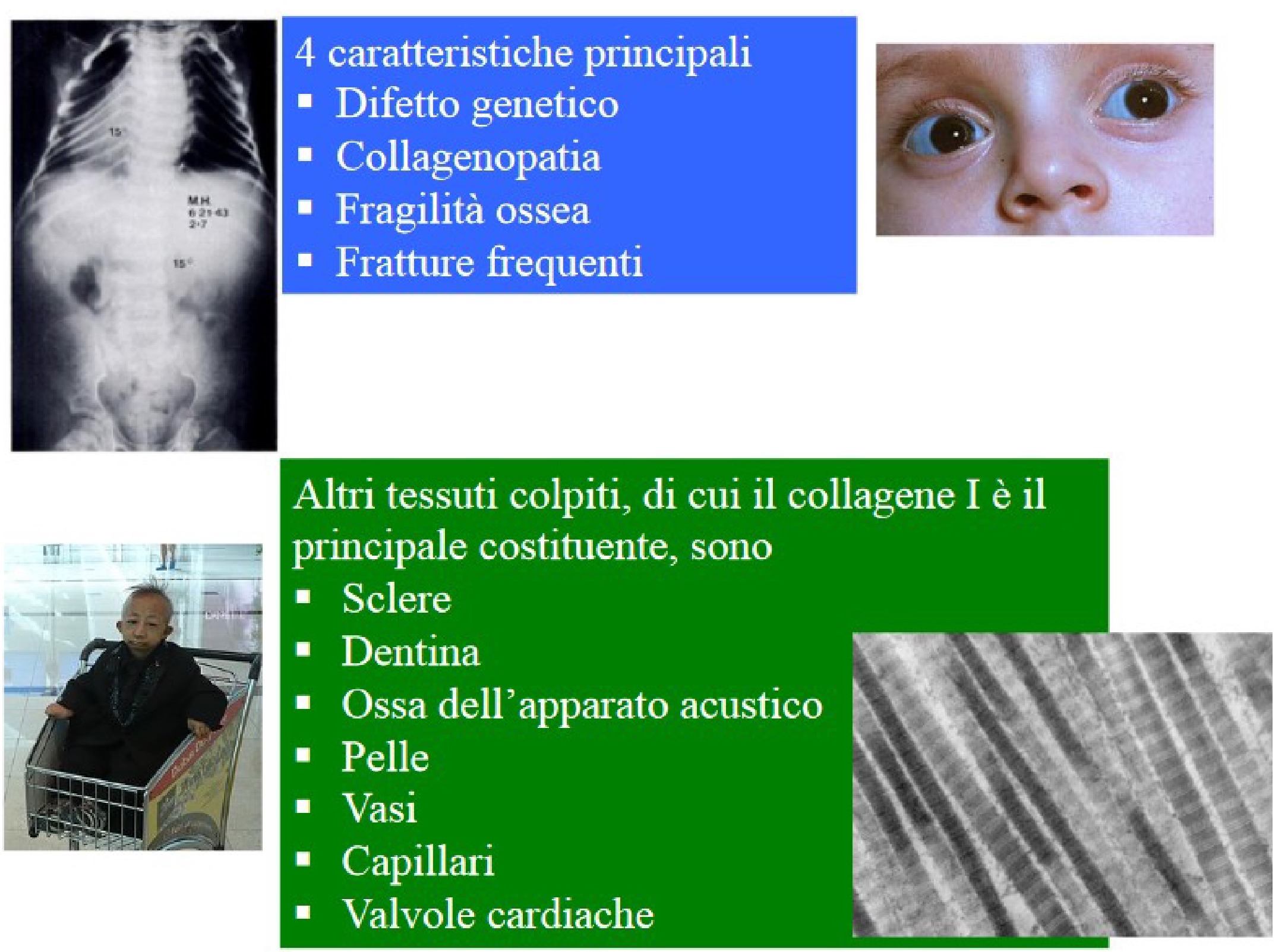
Figura 4
Caratteristiche cliniche dell'osteogenesi imperfetta
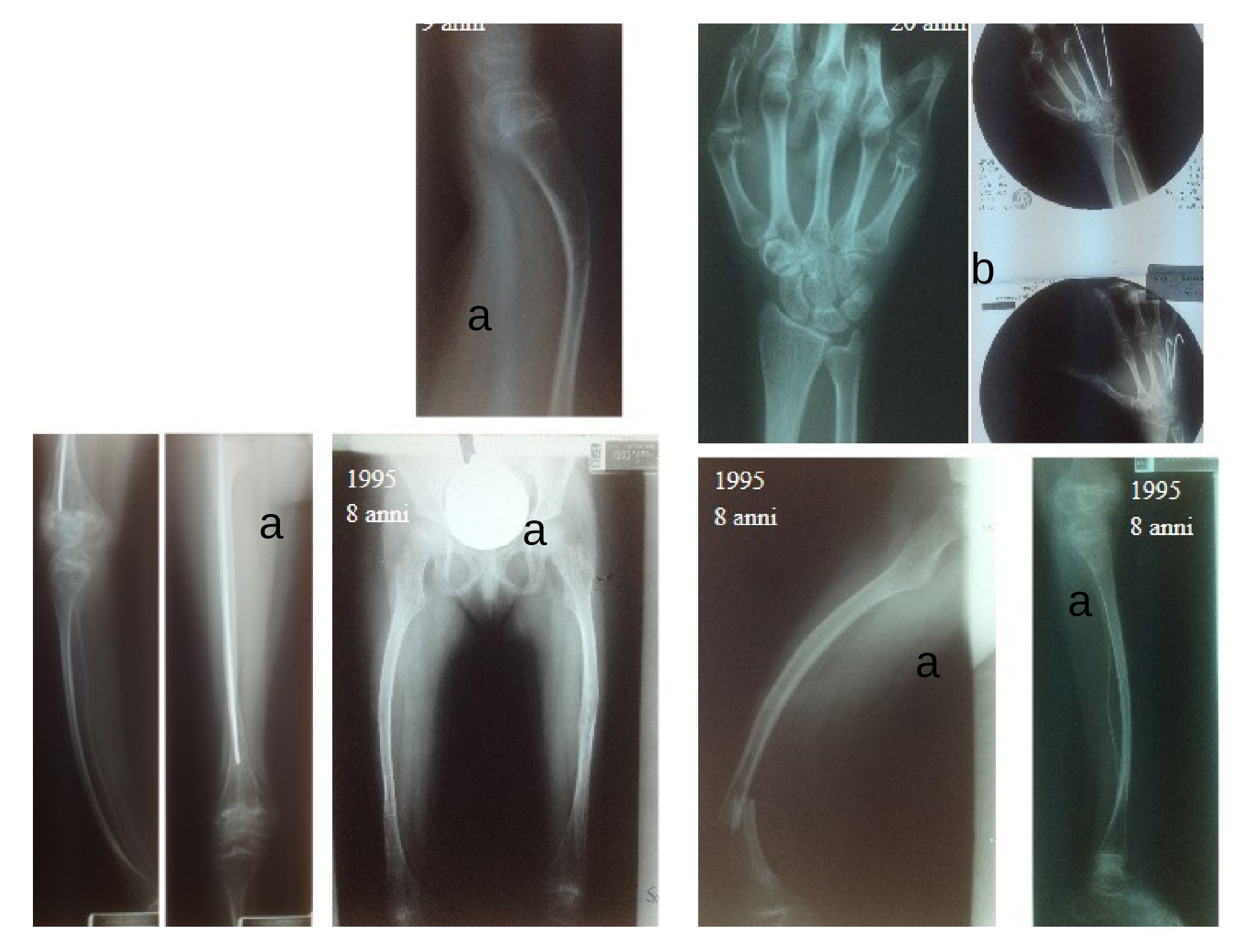
Figura 5
Ragazza con osteogenesi imperfetta di tipo III (trattata chirurgicamente per scoliosi a 15 anni), trattata chirurgicamente più volte in età pediatrica e adulta alle mani e agli arti inferiori.
Incidenza: relativamente frequente (0.4/10mila), di cui la metà è costituita dal tipo II.
Clinica: fratture spontanee, ipoplasia toracica, ipomineralizzazione, ossa deformate (incurvatura). Interessamento molto severo del rachide, con scoliosi dal 30% al 90% dei casi (aumenta con l’aumentare della severità della patologia). L’alta prevalenza di scoliosi è a sostegno della teoria che collega la collagenopatia con iperlassità dei legamenti della colonna e deformità a carico dei corpi vertebrali e frequenti micro-fratture vertebrali da fragilità, al danno a carico delle cartilagini di accrescimento, al verificarsi di fratture e deformità costali, oltre all'impossibilità di deambulare correttamente e ai problemi legati alla dentinogenesi imperfetta. La scoliosi è meno frequente prima dei 6 anni, ma poi va spesso in rapida progressione. Se la progressione della curva scoliotica non viene arrestata tempestivamente, la deformità può evolvere verso valori molto elevati (oltre i 90°) (fig 6), causando riduzione della funzionalità polmonare e sviluppo di cuore polmonare, con esito infausto.
Figura 6
Radiografie in ortostatismo di ragazza di 15 anni con osteogenesi imperfetta di tipo III, con severa scoliosi, precedentemente trattata con bisfosfonati e con ortesi bassa: pre-operatorie antero-posteriore (a) e latero-laterale (b); antero-posteriore (c) e latero-laterale (d), tre mesi dopo correzione ed artrodesi posteriore strumentata mediante viti peduncolari.
Diagnosi: può essere effettuata in fase pre-natale (nei casi gravi), clinicamente, radiograficamente o mediante esame biochimico o genetico.
Trattamento medico: bisfosfonati, anche in pazienti di età < 2 anni (tab 2-5).
Trattamento ortesico (busto ortopedico o corsetto in gesso): è controverso, perchè può causare deformità costali severe, con aggravamento secondario della scoliosi (è sicuramente controindicato un corsetto alto con mentoniera, che può causare malocclusione dentale e piaghe da decubito).
Trattamento chirurgico: oltre al trapianto di midollo osseo per aumentare la densità degli osteoblasti, riducendo così la frequenza delle fratture, consiste nell’armatura delle ossa lunghe. Attualmente sono già in uso strumentazioni intelligenti (cioè allungabili) per la correzione della deformità ossea, intra-midollari per le ossa lunghe e sul rachide per la scoliosi (6,8,20-22). Sin dagli anni ‘70 e ’80 c‘è stata indicazione al trattamento chirurgico delle scoliosi severe, mediante artrodesi strumentata posteriore, in particolare nei casi in cui la scoliosi mostra una rapida evoluzione in età precoce e il trattamento conservativo non ha successo. L'alta incidenza di complicanze, dovute alle difficoltà di ancoraggio dello strumentario per la ridotta resistenza meccanica del tessuto osseo, ha reso prudente negli anni l’approccio chirurgico alle deformità della colonna. Solo recentemente, grazie all’evoluzione degli strumentari (fig 6) e delle tecniche chirurgiche e anestesiologiche, gli interventi per scoliosi in questa patologia sono diventati meno rischiosi e con soddisfacente correzione intra-operatoria (6,23-26).
Ipofosfatasia
Genetica: mutazione del gene codificante per la fosfatasi alcalina. Ne esistono tre tipi, il III diagnosticabile in utero e con prognosi peggiore.
Incidenza: molto rara.
Clinica: micromelia, ipoplasia toracica, ipomineralizzazione, idrope ed edema nei casi diagnosticati precocemente.
SRPS (Short Rib Polydactyl Syndrome)
Eziologia: sconosciuta.
Incidenza: molto rara.
Clinica: se ne riconoscono due forme, entrambe con le stesse caratteristiche ecografiche, spesso associate a cardiopatia congenita. Micromelia, ipoplasia toracica, esadattilia.
Esito: letale nel 100% dei casi.
Displasia camptomelica
Genetica: mutazione del gene codificante la proteina SRY-box 9, presente nel testicolo e nello scheletro.
Incidenza: rara (0.2/10mila nati).
Clinica: rizomelia, tibia e femore incurvati, ipoplasia delle scapole, micrognazia, sex reversal nei maschi.
Esito: quasi sempre letale.
DISPLASIE SCHELETRICHE NON LETALI
Displasia distrofica
Genetica: mutazione del gene DTDST.
Incidenza: molto rara.
Clinica: deformità posturali, micrognazia, contratture degli arti (pollice da “autostoppista”).
Esito: letale nel 25% dei casi, grave handicap fisico per le contratture e la cifoscoliosi in coloro che sopravvivono.
Acondroplasia.
Genetica: mutazione del gene codificante per Fibroblast Growth Factor Receptor. Se ne distinguono due sottotipi: omozigote ed eterozigote.
Incidenza: relativamente frequente (1/10mila nati).
Clinica: rizomelia, tendenza alla macrocrania, naso insellato, mano a tridente.
Esito: sopravvivenza e performance mentale nella norma, problematiche ortopediche e polmonari a lungo termine.
Distrofia toracica asfissiante
Eziologia: sconosciuta.
Incidenza: rara.
Clinica: ipoplasia toracica, anomalie renali, rizomelia moderata.
Esito: letale nel 60% dei casi per ipoplasia polmonare.
Distrofia condro-ectodermica
Genetica: mutazione locus 4p16 che porta ad anomalia del tessuto di accrescimento cartilagineo tra epifisi e diafisi e a displasia dei tessuti di origine ectodermica.
Incidenza: molto rara (1/60mila nati).
Clinica: acro-mesomelia, ipoplasia toracica di media entità, esadattilia post-assiale, soprattutto delle mani, cardiopatia congenita (difetto inter-atriale).
Esito: letale nel 50% dei casi.
Distrofia metatropica
Eziologia: mutazione nel gene TRPV4, che codifica per un canale cationico permeabile al Ca2+ presente in diversi tessuti. Le mutazioni possono provocare un aumento del calcio nei condrociti e, di conseguenza, un'alterazione dell'ossificazione endo-condrale ed i segni clinici.
Incidenza: non nota (ad oggi sono stati descritti circa 81 casi) (1).
Clinica: displasia spondilo-epimetafisaria, caratterizzata da torace lungo e arti corti nel periodo neonatale, successiva cifo-scoliosi grave e progressiva, che esita in inversione delle proporzioni nell'infanzia (torace corto e arti lunghi) e statura finale bassa nell'età adulta. Lo spettro fenotipico è variabile e si associa a forme gravi con esito letale "in utero", o immediatamente dopo la nascita, e a forme che presentano solo lievi alterazioni scheletriche.
Diagnosi: si basa sui segni clinici e radiologici. Questi variano a seconda dell'età e comprendono diafisi corte con metafisi larghe, significativa plati-spondilia, calcificazione precoce della cartilagine ioide e cricoide, bacino a forma di alabarda, grave ipoplasia della parte anteriore delle prime vertebre cervicali, con anomalie e aspetto squadrato delle ossa del calcagno. I test molecolari possono identificare le mutazioni diTRPV4, confermando la diagnosi.
Esito: la prognosi varia a seconda della gravità della malattia. L'attesa di vita non è di solito compromessa, a meno che non vi siano complicazioni respiratorie.
CONCLUSIONI
Il percorso diagnostico delle displasie scheletriche è fondamentalmente clinico-radiologico: la valutazione accurata dei segni clinici e radiologici può consentire almeno l’inquadramento in uno dei 42 gruppi o famiglie e indirizzare a eventuali indagini genetiche mirate.
Negli ultimi vent’anni sono stati compiuti notevoli progressi nella comprensione dei disturbi correlati a mutazioni del recettore FGF-R3 e dei meccanismi patogenetici alla base di tali disturbi, in modo da delineare strategie terapeutiche efficaci nel trattamento dei difetti di crescita ossea associati a questo tipo di mutazioni. Sebbene ci sia stato un certo successo nello sviluppo di nuove terapie, il miglioramento ulteriore del trattamento di bambini e adulti affetti rappresenta tuttora una sfida affascinante. Ci sono diverse nuove strategie terapeutiche che potranno essere prese in considerazione in futuro. Inoltre, sarà importante studiare il potenziale sinergismo di due o più inibitori farmacologici dell’FGF-R3 o della via di segnale a valle, che potrebbero aumentare l’efficacia del trattamento. L’eterogeneità clinica e allelica di diverse condizioni, in relazione a mutazioni in domain diversi di uno stesso gene e soprattutto in malattie estremamente rare, può complicare l’iter diagnostico. L’attuale livello di conoscenze ha reso possibile la diagnosi con diversi criteri clinici, radiologici e molecolari della maggior parte delle 461 displasie scheletriche attualmente note. In questi casi, per giungere alla diagnosi è indispensabile ricorrere a un’indagine molecolare con sequenziamento completo dell'esoma, che consente di definire la diagnosi nella quasi totalità dei casi (92%). Nello stesso tempo sono molto migliorate le condizioni di vita dei pazienti grazie ai progressi in campo terapeutico, sia chirurgico, per prevenire le deformità, sia farmacologico con il ricorso a farmaci nuovi o già in uso per altre indicazioni, sia con la terapia enzimatica sostitutiva (ad es. in alcune mucopolisaccaridosi), ma soprattutto con terapie specifiche (trial già conclusi o in itinere nei casi di acondroplasia).
BIBLIOGRAFIA
- Scarano G, Falco M, Scarano F, et al. Bassa statura e sindromi rare: le displasie scheletriche. Archivio MR 2020 - 2.
- Tsang KY, Tsang SW, Chan D, Cheah KSE. The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Def Res (Part C) 2014, 102: 52–73.
- Spranger J, Maroteaux P. The lethal osteochondrodysplasias. Adv Hum Genet 1990, 19: 1-103, 331-2.
- Stanescu V, Stanescu R, Maroteaux P. Pathogenic mechanisms in osteochondrodysplasias. J Bone Joint Surg Am 1984, 66: 817-36.
- Mortier GR, Cohn DH, Cormier-Daire V, et al. Nosology and classification of genetic skeletal of genetic skeletal disorders: 2019 revision. Am J Med Genet 2019, 179A: 2393-419.
- Bonafè L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet Part A 2015, 167: 2869-92.
- Hoover-Fong J, Scott CI, Jones MC; Committee on Genetics. Health supervision for people with achondroplasia. Pediatrics 2020, 145: e20201010.
- Schiedel F, Rödl R. Lower limb lengthening in patients with disproportionate short stature with achondroplasia: a systematic review of the last 20 years. Disabil Rehabil 2012, 34: 982-7.
- Donaldson J, Aftab S, Bradish C. Achondroplasia and limb lengthening: results in a UK cohort and review of the literature. J Orthop 2015, 12: 31-4.
- Kim SJ, Pierce W, Sabharwal S. The etiology of short stature affects the clinical outcome of lower limb lengthening using external fixation. A systematic review of 18 trials involving 547 patients. Acta Orthop 2014, 85: 181-6.
- Semler O, Rehberg M, Mehdiani N, et al. Current and emerging therapeutic options for the management of rare skeletal diseases. Paediatr Drugs 2019, 21: 95-106.
- Wang J, Zhou J, Cheng CM, et al. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol 2004, 180: 247–55.
- Gorbenko O, Ovcharenko G, Klymenko T, et al. Generation of monoclonal antibody targeting fibroblast growth factor receptor 3. Hybridoma (Larchmt) 2009, 28: 295-300.
- Gust KM, McConkey DJ, Awrey S, et al. Fibroblast growth factor receptor 3 is a rational therapeutic target in bladder cancer. Mol Cancer Ther 2013, 12: 1245-54.
- Liu Z, Lavine KJ, Hung IH, Ornitz DM. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev Biol 2007, 302: 80-91.
- Garcia S, Dirat B, Tognacci T, et al. Postnatal soluble FGFR3 therapy rescues achondroplasia symptoms and restores bone growth in mice. Sci Transl Med 2013, 5: 203ra124.
- Breinholt VM, Rasmussen CE, Mygind PH, et al. TransCon CNP, a sustained-release C-type natriuretic peptide prodrug, a potentially safe and efficacious new therapeutic modality for the treatment of comorbidities associated with fibroblast growth factor receptor 3-related skeletal dysplasias. J Pharmacol Exp Ther 2019, 370: 459-71.
- Argente J, Martos Moreno GA. Skeletal dysplasias: new medical treatments. An Pediatr (Barc) 2016, 85: 1-3.
- Greggi T. Esperienza e stabilizzazione vertebrale nell'osteogenesi imperfetta. L'osteogenesi imperfetta: il percorso diagnostico, terapeutico e assistenziale dal bambino all’adulto. Congresso Nazionale Associazione Italiana Osteogenesi Imperfetta Onlus - Milano 2-5 maggio 2013.
- Wallace MJ, Kruse RW, Shah SA. The spine in patients with osteogenesis imperfecta. J Am Acad Orthop Surg 2017, 25: 100-9.
- Keuning MC, Leeuwerke SJG, van Dijk PR, et al. Prevalence of scoliosis and impaired pulmonary function in patients with type III osteogenesis imperfecta. Eur Spine J 2022, 31: 2295-300.
- Burnei G, Vlad C, Georgescu I, et al. Osteogenesis imperfecta: diagnosis and treatment. J Am Acad Orthop Surg 2008, 16: 356-66.
- Paley D. PRECICE intramedullary limb lengthening system. Expert Rev Med Devices 2015, 12: 231-49.
- Piantoni L, Noel MA, Francheri Wilson IA, et al. Surgical treatment with pedicle screws of scoliosis associated with osteogenesis imperfecta in children. Spine Deform 2017, 5: 360-5.
- Karlin LI, McClung A, Johnston CE, et al; Children’s Spine Study Group. The growth-friendly surgical treatment of scoliosis in children with osteogenesis imperfecta using distraction-based instrumentation. Spine Deform 2021, 9: 263-74.
- Gardner A, Sahota J, Dong H, et al. The use of magnetically controlled growing rods in paediatric osteogenesis imperfecta. J Surg Case Rep 2018, 2018: rjy043.
Introduzione alle malattie rare
Elisa Parolo, Francesca Dassie, Francesca Favaretto, Gabriella Milan, Pietro Maffei
Azienda Ospedaliera di Padova, Clinica Medica 3^, DIMED
(aggiornato al 21 luglio 2016)
Introduzione
Le malattie rare sono patologie che possono incidere fortemente su sopravvivenza e qualità di vita di chi ne è affetto e vengono generalmente definite sulla base delle caratteristiche epidemiologiche di numerosità. Secondo l’Unione Europea (UE), le malattie rare sono patologie cronicamente debilitanti o che compromettono la sopravvivenza, caratterizzate da una bassa prevalenza, ovvero che colpiscono non più di 5 persone su 10.000 (0.05% della popolazione) (1). Vi è un paradosso tra la rarità di tali malattie e la numerosità dei casi: la Commissione Europea ha, infatti, stimato che esistano tra le 5.000 e le 8.000 malattie rare, che colpiscono il 6-8% della popolazione, ossia circa 27-36 milioni di persone nella sola UE (2). Le malattie rare costituiscono quindi nel loro insieme un rilevante problema di salute pubblica, tale per cui negli anni sono stati fatti molti sforzi per favorire l’assistenza e la ricerca in questo settore, anche adeguando la legislazione comunitaria.
A partire dal 1999, si è registrato un crescente interesse in questo ambito e il regolamento CE n° 141/2000 del Parlamento Europeo e del Consiglio, che era stato inizialmente pensato per identificare i farmaci orfani ed incentivarne lo sviluppo e la commercializzazione, nei fatti è diventato la base di partenza per definire il problema “malattia rara” e per portarlo all’attenzione dell’opinione pubblica. In seguito, nel 2009, il Consiglio Europeo ha promulgato una direttiva per l’adozione di un piano nazionale di sorveglianza sulle malattie rare (3). Nello stesso anno fu istituita la commissione EUCERD (European Committee of Experts on Rare Diseases), costituita da esperti con l’obiettivo di guidare l’UE e gli stati membri alla pianificazione più adeguata, per migliorare la politica sanitaria e sociale nell’ambito delle malattie rare. Nel 2013 questa commissione è stata sostituita dall’EGRD (Expert Group on Rare Diseases), che ha comunque perseguito gli stessi obiettivi (4). Appare quindi evidente il forte impegno legislativo da parte della UE e dei paesi membri per affrontare il problema e adottare politiche sanitarie atte alla miglior presa in carico dei pazienti (5).
Le malattie rare sono molto eterogenee per età di insorgenza, ezio-patogenesi e sintomatologia; possono interessare uno o più organi e apparati. La maggior parte riconosce una causa genetica. Tuttavia, la classificazione risulta non semplice, comprendendo anche patologie del sistema immunitario, errori congeniti del metabolismo, malattie endocrinologiche e neoplasie rare (6). Nonostante la spiccata eterogeneità, esse condividono tuttavia alcune problematiche gestionali comuni, che ne rendono opportuno il raggruppamento, allo scopo di programmare specifici interventi di sanità pubblica.
Difficoltà diagnostiche
Le malattie rare sono frequentemente sotto-stimate, non diagnosticate o erroneamente diagnosticate per vari motivi:
- l’esordio può avvenire con sintomi aspecifici, subdoli, che possono risultare sovrapponibili a quelli di altre patologie, per cui può risultare difficile discriminare una patologia rara dall’altra o da patologie più frequenti;
- alcune condizioni, relativamente comuni, possono mascherare la presenza di una malattia rara;
- l’eterogeneità dello spettro fenotipico con cui si può manifestare la malattia stessa;
- la condizione stessa di rarità fa sì che la malattia venga poco studiata e quindi difficilmente riconosciuta.
Tutti questi aspetti costituiscono un ulteriore ostacolo per la diagnosi e molto spesso il tempo di latenza tra l’esordio della patologia e la diagnosi può risultare molto lungo, incidendo negativamente sulla prognosi del paziente, nonché sulla sua qualità di vita. Inoltre, una diagnosi errata può implicare la mancanza di cure o la somministrazione di cure inappropriate e potenzialmente dannose per il paziente.
Per superare queste difficoltà, si sono elaborate diverse strategie, in primis la diffusione e condivisione delle informazioni consultabili liberamente. Tra le varie iniziative, ricordiamo RD-Connect (Rare Disease-Connect), nata nel novembre 2012 sotto il patrocinio dell’IRDiRC (International Rare Diseases Research Consortium), un consorzio multinazionale che ha come obiettivo comune l’identificazione delle malattie rare e lo sviluppo di nuove terapie. RD-Connect rappresenta un progetto globale che ha vari obiettivi, come lo sviluppo di una piattaforma informatica integrata che possa mettere a disposizione, e quindi a confronto, dati genetici, clinici, derivati da strumenti bio-informatici o da registri nazionali, allo scopo di aumentare il bagaglio di nozioni e quindi di esperienza nel campo delle malattie rare (7).
Per rendere maggiormente fruibili le informazioni, sono nati molti altri strumenti, come ad esempio ORPHANET, sito web che offre vari servizi, fra cui un elenco dettagliato e una classificazione delle malattie rare, linee guida, un elenco aggiornato dei farmaci orfani e dei centri di riferimento, nonché le informazioni derivanti dalle sperimentazioni e dai progetti di ricerca. ORPHANET Italia è il sito italiano su cui si possono trovare notizie, eventi e documenti sulle malattie rare e i farmaci orfani rilevanti a livello nazionale.
Ricordiamo infine il progetto EUROPLAN (European Project for Rare Diseases National Plans Development), cofinanziato dalla Commissione Europea e coordinato in Italia dal Centro Nazionale delle Malattie Rare (CNMR), nato con l’intento di promuovere piani nazionali per affrontare il problema delle malattie rare, da una parte agendo con strategie comuni e dall’altro tenendo conto delle indicazioni date dalla Commissione Europea (8).
Opzioni terapeutiche
Sono generalmente limitate, tuttavia la progressiva presa di coscienza dell’esistenza di tali patologie e gli sforzi fatti per la loro gestione hanno portato a incentivare la ricerca per lo sviluppo di terapie adeguate.
Un fattore determinante è dato dal fatto che l’investimento nella produzione e nella commercializzazione di farmaci di uso limitato, fa sì che essi non siano remunerativi dal punto di vista economico. Questo fattore limita la ricerca e la sperimentazione e tutto ciò porta a indicare come “orfani” questi farmaci. Il farmaco orfano si definisce, infatti, come una sostanza potenzialmente utile a trattare una malattia rara, ma che non ha un mercato sufficiente per ripagare le spese del suo sviluppo; di conseguenza, non vi è interesse, da parte delle case farmaceutiche, ad investire su di esso (9). Per queste ragioni si è palesata l’esigenza di una specifica legislazione che sostenga la ricerca e lo sviluppo di farmaci orfani e renda sostenibile il costo della loro realizzazione.
Nel 1983 gli Stati Uniti per primi hanno emanato una legislazione sui farmaci orfani (10). Negli anni seguenti, molte nazioni e le istituzioni europee e nazionali hanno varato leggi specifiche, atte a incentivare la ricerca e la sperimentazione nel settore dei farmaci orfani o a regolamentare l’erogazione di quelli già disponibili. A livello europeo, nel 1995 è stata istituita l’EMA (European Medicines Agency), agenzia che si occupa della valutazione dei farmaci prima della loro immissione nel mercato, e della loro sicurezza anche attraverso uno stretto monitoraggio. È stato inoltre istituito il COMP (Committee for Orphan Medicinal Products), che si occupa della presa in carico delle richieste di valutazione e designazione dei farmaci orfani, attraverso parametri ben precisi, basati essenzialmente su criteri di prevalenza ed economici. ll Parlamento Europeo e il Consiglio d’Europa hanno poi promulgato nel 2000 una normativa (CE n° 141/2000) atta a incentivare sviluppo e commercializzazione dei farmaci orfani, stabilendo precisi criteri di designazione. Tuttavia, le modalità di designazione e approvazione dei farmaci orfani, nonché gli incentivi per il loro sviluppo e la normativa per la loro erogazione differiscono ancora sensibilmente tra le varie nazioni (11). La fase di commercializzazione dei farmaci orfani è controllata dal CHMP (Committee for Human Medicinal Products), comitato che autorizza la disponibilità sul mercato dei suddetti farmaci seguendo precise linee guida e utilizzando le informazioni derivanti da studi clinici adeguatamente programmati e condotti (12). Il percorso per l’autorizzazione e la commercializzazione di un farmaco orfano è quindi lungo e a volte non scevro da ostacoli (13); una volta giunto a termine, è la Comunità Europea stessa che si occupa della sua regolamentazione finale.
L’Italia segue la normativa europea e le normative nazionali applicate dall’AIFA (Agenzia Italiana del Farmaco), secondo la quale la disponibilità dei farmaci orfani è determinata sia dalle AIC, ovvero le Autorizzazioni all’Immissione in Commercio dei singoli medicinali pubblicate nella Gazzetta Ufficiale (GU), sia dalle liste previste dalla Legge 648 (14). Quest’ultima legge consente di erogare il farmaco a carico del Sistema Sanitario Nazionale (SSN) quando non vi è nessun’altra opzione terapeutica valida; questo può avvenire dopo la valutazione positiva della Commissione Tecnico-Scientifica dell’AIFA, con successiva pubblicazione in GU dell’allegato in cui vengono precisate le modalità di impiego e i parametri da monitorare ai fini della farmaco-vigilanza. Sul sito dell’AIFA è disponibile l’elenco dei farmaci orfani e la normativa di riferimento. La legge n° 79, pubblicata in GU nel maggio 2014, parla di “disposizioni urgenti in materia di disciplina degli stupefacenti e sostanze psicotrope, prevenzione, cura e riabilitazione dei relativi stati di tossico-dipendenza, nonché di impiego di medicinali meno onerosi da parte del SSN”. È quindi articolata in due diverse disposizioni, la prima che si riferisce alle disposizioni sui farmaci stupefacenti, la seconda che invece si occupa della revisione della disciplina dell’uso off-label dei farmaci (15). Questa legge prevede, infatti, che, previa valutazione dell’AIFA, possano essere erogati a carico del SSN farmaci impiegati per un’indicazione terapeutica diversa da quella autorizzata (uso off-label), se tale indicazione risulta conforme alle ricerche condotte nella comunità medico-scientifica nazionale e internazionale, rispettando i parametri di appropriatezza ed economicità (16).
Non tutti i farmaci impiegati nella cura delle malattie rare si possono definire orfani, infatti ve ne sono alcuni che, inizialmente concepiti per una patologia diffusa, si sono scoperti utili al controllo di una patologia rara; la loro prescrizione ed erogazione viene fatta solo per casi selezionati, ed è regolata da procedure specifiche (17).
Percorsi assistenziali
La condizione di rarità implica che sia poco conosciuta la fisio-patologia di molte malattie, e ciò ha rallentato lo sviluppo di percorsi adeguati, con potenziali disparità di trattamento rispetto alle patologie più comuni. In linea generale, la risposta assistenziale dovrebbe essere sempre di elevata qualità, per cui è imperativo offrire ai malati un’assistenza ultra-specialistica c/o strutture adeguate con operatori sanitari esperti, in grado di fornire ai pazienti le risposte che soddisfino i loro bisogni di salute.
Con il DM n. 279 del 18.05.2001 è stata istituita la Rete Nazionale dedicata alle malattie rare, con lo scopo di creare un organo istituzionale in grado di sviluppare azioni di prevenzione e sorveglianza, migliorare gli interventi atti alla diagnostica e alla terapia, stabilire le esenzioni per i servizi compresi nei LEA (Livelli Essenziali di Assistenza) ed infine incentivare l’informazione e favorire la formazione degli operatori sanitari (18). La Rete Nazionale è sostanzialmente costituita da tutte le strutture e i servizi delle Regioni, le quali, a loro volta, hanno individuato appositi centri di diagnosi e cura accreditati, preferibilmente ospedalieri, sulla base dell’esperienza maturata nella gestione delle malattie rare e sulla dotazione di strutture di supporto e servizi complementari. Questi centri si occupano di vari aspetti: in primis la formulazione della diagnosi di malattia rara erogando le prestazioni necessarie, comprese le indagini genetiche, in regime di esenzione; poi, una volta posta la diagnosi, si occupano della redazione del Piano Terapeutico Personalizzato (PTP) e dell’erogazione delle relative cure, sempre in regime di esenzione. I centri compresi nella Rete operano secondo protocolli clinici specifici e cooperano con i servizi territoriali e i medici di medicina generale per la presa in carico dei pazienti e la gestione diagnostico-terapeutica. È stato altresì istituito un numero verde per le malattie rare (TVMR 800.89.69.49), raggiungibile gratuitamente da tutta Italia ed anche dai cellulari, al fine di erogare informazioni sulle malattie rare e sulla rete nazionale. Il DM 279/2001, ha anche previsto l’istituzione di un Registro delle Malattie Rare, che possa inglobare tutti i dati dei registri regionali, allo scopo di raccogliere quante più informazioni possibili sia epidemiologiche che cliniche. Mediante indagini epidemiologiche mirate si cerca così di chiarire le dimensioni del problema, definire la prevalenza e/o l’incidenza delle varie patologie, identificare i possibili fattori di rischio, per impostare un eventuale programma di prevenzione, migliorare la definizione dei criteri diagnostici per una diagnosi più tempestiva e adeguata, sostenere la ricerca clinica con il maggior numero possibile di dati certificati (19).
Un contributo fondamentale alla creazione della Rete è stato dato fin dal 2000 dalla “Unità di Malattie Rare”, all’interno dell’Istituto Superiore di Sanità (ISS), che ha costituito il nucleo embrionale del CNMR, istituito ufficialmente con Decreto del Presidente dell’ISS nel 2008 (GU n° 157 del 7/7/2008), con la missione di svolgere attività di ricerca, consulenza e documentazione sulle malattie rare e i farmaci orfani, finalizzata alla prevenzione, trattamento e sorveglianza delle stesse (20). Il CNMR rappresenta l’Italia nel comitato per i farmaci orfani (COMP) istituito presso l’EMA e in generale persegue i seguenti obiettivi:
- promozione dell’attività di ricerca sulle malattie rare, utilizzando le tecnologie più avanzate;
- coordinamento del progetto EUROPLAN citato in precedenza;
- elaborazione di test genetici, con l’identificazione di marcatori diagnostici e prognostici, validazione e controllo di qualità dei test genetici;
- prevenzione, sorveglianza (registro delle malattie rare), formazione continua degli specialisti e divulgazione dell’informazione;
- erogazione di farmaci orfani;
- promozione del processo di collaborazione europea e internazionale sulle malattie rare;
- promozione della medicina narrativa; in collaborazione con le associazioni dei pazienti, viene data la possibilità per i soggetti affetti di raccontare la loro storia attraverso una forma artistica in un concorso chiamato “Il volo di Pegaso”, con successiva premiazione dei migliori lavori durante la Giornata delle Malattie Rare, che si svolge solitamente l’ultimo giorno di febbraio di ogni anno.
Classificazione e codifica
È molto importante riuscire a tradurre la diagnosi di una malattia in un codice specifico, allo scopo di identificarla e renderla rintracciabile nei sistemi informativi sanitari. Tutto questo per garantire flussi di informazioni, che fungano da base per la produzione di statistiche di dati di morbilità, mortalità, efficacia e qualità dei sistemi sanitari. Tutto ciò si concretizza in uno strumento necessario per approntare misure appropriate di sanità pubblica. Tuttavia, la classificazione e codifica delle malattie rare si è rivelata essere molto complessa; questo rappresenta una problematica di rilievo, poiché codici inappropriati possono comportare erronee analisi statistiche e quindi rendere i risultati poco fruibili o confondenti.
Attualmente la classificazione viene effettuata con il sistema dell’International Classification of Diseases (ICD), coordinato dall’OMS (Organizzazione Mondiale della Sanità). Tuttavia, questo sistema non è adeguato per molte malattie rare, dato che poche di esse hanno codici univoci specifici, e molte sono codificate all’interno di gruppi eterogenei. Negli anni si sono compiuti numerosi sforzi per migliorare il sistema ICD, e già nel 2004 la Commissione Europea istituiva un gruppo di lavoro coordinato dall’EUCERD e in collaborazione con l’OMS, per la revisione della codifica e classificazione delle Malattie Rare, coinvolgendo più esperti (clinici, epidemiologi, statistici). Questo lavoro è stato diretto non solo alla revisione dell’ICD attualmente in uso (ICD-9, ICD-10 e 11), ma anche alla realizzazione di un nuovo sistema di classificazione capace di migliorare ulteriormente l’identificazione delle malattie rare. Nella versione attuale dell’ICD-11 (beta version) sono classificate circa 5.000 malattie rare (21). A questo gruppo di lavoro collabora attivamente anche il CNMR.
Associazioni pazienti
I pazienti e i familiari devono spesso affrontare un’esperienza dolorosa, legata sia alla malattia che alla condizione di rarità, con molte delle difficoltà precedentemente elencate. Senza contare che molti vivono la patologia genetica rara come una “colpa”, poiché non identificano la malattia in qualcosa che ha colpito l’individuo, ma identificano l’individuo come malattia, e secondo un sottile meccanismo psicologico il malato viene visto come un errore e la famiglia si sente colpevole per tutto questo. Ecco perché è necessaria un’adeguata informazione sulla malattia rara, che non deve essere vissuta come una condizione di diversità. Negli anni è emersa la necessità, da parte dei pazienti e delle famiglie, di creare una comunicazione e una condivisione dell’esperienza di malattia, allo scopo di evitare situazioni di solitudine e garantire sostegno durante tutte le tappe della malattia stessa. Sono nate così varie associazioni, che nel tempo hanno guadagnato un ruolo determinante sia nel produrre conoscenza della malattia rara in quanto tale, sia per quanto riguarda la presa in carico di situazioni spesso difficili causate dalla convivenza con la malattia stessa. Inoltre, la condivisione delle esperienze crea una rete di solidarietà, che permette di affrontare anche il doloroso percorso di accompagnamento dei malati verso l’exitus.
Tra le varie associazioni italiane ricordiamo UNIAMO, federazione italiana malattie rare, fondata nel 1999, che ad oggi comprende circa più di 100 associazioni di affetti e loro familiari, per oltre 600 patologie rare rappresentate. UNIAMO fa parte di EURORDIS (European Organisation for Rare Diseases), federazione europea non governativa, che comprende 705 associazioni di affetti da malattia rara in 63 paesi, per almeno 4.000 patologie rare rappresentate. Negli Stati Uniti è stato invece creato NORD (National Organization for Rare Disorders), associazione che cerca di riunire gli affetti da malattie rare nei paesi membri degli USA. Queste sono solo alcune delle varie associazioni di pazienti, che tutte hanno lo scopo di dare voce e visibilità al mondo delle malattie rare, e restituire così dignità ai soggetti affetti.
Ricordiamo infine che la stessa Comunità Europea ha favorito il coinvolgimento dei rappresentanti delle associazioni pazienti come parte attiva nella pianificazione delle politiche sanitarie, nella ricerca e nella gestione delle risorse.
Bibliografia
- European Parliament and the Council. Regulation (EC) 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. Official Journal 2000, L 18: 1-5.
- Ministero della Salute. Piano Nazionale delle Malattie Rare 2013-6.
- European Council. Recommendation of 8 June 2009 on European Action in the field of rare diseases. Official Journal C 151: 7-10.
- European Commission. Commission Expert Group on Rare Diseases.
- Rodwell C, Aymé S. Rare disease policies to improve care for patients in Europe. Biochim Biophys Acta 2015, 1852: 2329-35.
- Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet 2008, 371: 2039-41.
- Thompson R, Johnston L, Taruscio D, et al. RD-Connect: an integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. J Gen Intern Med 2014, 29 suppl 3: 780-7.
- EUCERD and EUROPLAN. Joint report on initiatives and incentives in the field of rare diseases of the European Union Committee of experts on rare diseases; 2009.
- Van Weely S, Leufkens HGM. Background paper: orphan disease. In: Kaplan W, Laing R (Eds). Priority medicines for Europe and the world – a public health approach to innovation. Geneva: World Health Organization; 2004.
- US code of Federal Regulations (CFR), Title 21 Food and Drugs, Part 316 Orphan Drugs.
- Gammie T, Lu CY, Babar ZU. Access to orphan drugs: a comprehensive review of legislations, regulations and policies in 35 countries. PLoS One 2015, 10: e0140002.
- European Medicines Agency. The guidelines on clinical trials in small population; Reflection Paper on methodological issues in confirmatory clinical trials planned with an adaptative design. 2007.
- Haffner ME, Torrent–Farnell J, Maher PD. Does orphan drug legislation really answer the needs of patients? Lancet 2008, 371: 2041-4.
- Legge 648/96. Gazzetta Ufficiale n. 300, 23 dicembre 1996.
- Decreto Legge 17 febbraio 1998. Disposizioni urgenti in materia di sperimentazioni cliniche in campo oncologico e altre misure in materia sanitaria. Gazzetta Ufficiale 39, 17 febbraio 1998.
- Conversione in legge, con modificazioni, del DL 20 marzo 2014, n. 36, recante disposizioni urgenti in materia di disciplina degli stupefacenti e sostanze psicotrope, prevenzione, cura e riabilitazione dei relativi stati di tossicodipendenza, di cui al DPR 9 ottobre 1990, n. 309, nonchè di impiego di medicinali meno onerosi da parte del Servizio sanitario nazionale. Legge n. 79, 16 maggio 2014.
- Legge 8 aprile 1998, n. 94. Conversione in legge, con modificazioni, del Decreto Legge 17 febbraio 1998, n. 23, recante disposizioni urgenti in materia di sperimentazioni cliniche in campo oncologico e altre misure in materia sanitaria. Gazzetta Ufficiale 86, 14 aprile 1998.
- Taruscio D, Vittozzi L. The Italian approach to rare diseases and the action of the Italian National Centre for Rare Diseases. JPH 2009, 6: 267-71.
- Taruscio D, Kodra Y, Ferrari G, Vittozzi L; National Rare Diseases Registry Collaborating Group. The Italian National Rare Diseases Blood Transfus 2014, 12 suppl 3: s606-13.
- Gazzetta Ufficiale, Serie Generale, n.157 del 7/7/2008 (Decreto 26 giugno 2008 dell’ISS)
- World Health Organization, ICD-11 beta draft (accessed 5 december 2014).
Siti web:
- International Rare Diseases Research Consortium
- Orphanet
- Orphanet Italia
- European Project for Rare Diseases National Plan Development
- European Medicines Agency
- Agenzia Italiana del Farmaco
- Ministero della Salute
- Istituto Superiore di Sanità: Centro Nazionale Malattie Rare
- Federazione Italiana Malattie Rare
- Eurordis: rare diseases Europe
- NORD: National Organization for Rare Disorders
Sindromi complesse
Overview sulle sindromi complesse: quando sospettarle?
Maria Piccione & Martina Busè
Dipartimento di Scienze per la Promozione della Salute e Materno-Infantile “Giuseppe D’Alessandro”, Università degli Studi di Palermo
Le sindromi malformative complesse sono quadri clinici eterogenei, caratterizzati da malformazioni maggiori, note dismorfiche, anomalie dell’accrescimento e anomalie dello sviluppo psico-motorio e cognitivo.
Si tratta, in genere, di malattie rare, ovvero con una prevalenza alla nascita < 1/2000 nati vivi. Se considerate però globalmente, 1-2 neonati ogni 1000 nati vivi presenta una sindrome malformativa. Nella popolazione pediatrica italiana, la prevalenza dei pazienti sindromici è pari a 1/250.
A causa della loro rarità, le sindromi complesse sono poco conosciute e, spesso, non vengono diagnosticate o sono diagnosticate tardivamente, con conseguenze negative per il paziente e per la sua famiglia. Una diagnosi precisa e precoce, al contrario, consente di definire la prognosi del bambino e un programma di follow-up mirato, con una presa in carico multi-disciplinare e multi-specialistica, e di fornire una consulenza genetica alla famiglia del paziente (genitori, fratelli e sorelle, famiglia allargata). È quindi fondamentale che il pediatra non specialista sia in grado di riconoscere quegli elementi clinici cardine che consentono, non tanto di diagnosticare una sindrome complessa ma, almeno, di sospettarla in modo da attivare ulteriori approfondimenti.
Un corretto approccio diagnostico per il neonato/bambino con sospetta sindrome malformativa complessa comprende:
- accurata raccolta anamnestica;
- valutazione dei paramenti auxologici;
- analisi del fenotipo, valutando la presenza di note dismorfiche (anomalie minori) e malformazioni maggiori (a tal fine importante integrare l’esame clinico con screening malformativo ecografico ed eventuali indagini radiologiche e di imaging);
- valutazione dello sviluppo psico-motorio e cognitivo.
Anamnesi
L’anamnesi rappresenta il primo passo per un corretto approccio diagnostico.
Una corretta anamnesi familiare prevede la stesura di un albero genealogico di almeno tre generazioni, con informazioni circa eventuali aborti, morti intra-uterine, mortalità peri-natale, infantile e giovanile, malformazioni congenite isolate o complesse, ritardo psico-motorio, ritardo mentale, problemi neuro-funzionali, epilessia, disturbi dell’accrescimento ed eventuale consanguineità dei genitori.
L’anamnesi gravidica è volta a indagare circa la presenza di malattie materne, quali patologie autoimmuni, iperfenilalaninemia, diabete, infezioni contratte in gravidanza (TORCH), minacce di aborto o parto pretermine, eventuale uso di farmaci, assunzione di alcool e/o droghe, esposizione a radiazioni.
Per quanto riguarda l’anamnesi fetale, sarà importante ottenere informazioni relative a inizio e tipo di attività fetale, crescita fetale e malformazioni riscontrate tramite ecografie prenatali.
Nella raccolta dell’anamnesi peri-natale è necessario valutare i paramenti auxologici alla nascita in rapporto all’età gestazionale, la presenza di malformazioni maggiori e/o minori riscontrate alla nascita e/o di alterazioni neuro-motorie, quali ipotonia o ipertonia, difficoltà di suzione e alimentazione, convulsioni.
L’anamnesi fisiologica e quella patologica, infine, possono fornire ulteriori elementi per porre il sospetto diagnostico di una sindrome complessa.
Malformazioni maggiori
Le malformazioni maggiori sono quelle anomalie morfo-strutturali che determinano un’alterazione di una funzione, tale da necessitare di trattamento medico e/o chirurgico. La prevalenza alla nascita è elevata, pari al 2-4% dei nati vivi, ed esse costituiscono la terza causa di ospedalizzazione nei primi 4 anni di vita in Italia. È chiaro che non tutte le malformazioni maggiori sono parte di un quadro sindromico complesso: la maggior parte è, infatti, un difetto congenito isolato. Ciononostante, la percentuale di pazienti con una malformazione maggiore affetti da una sindrome malformativa complessa è elevata, e cresce in caso di presenza di più di una malformazione maggiore o della contemporanea presenza di anomalie minori, anomalie dell’accrescimento e/o di sviluppo cognitivo e psico-motorio. Per tale motivo, il riscontro di una malformazione maggiore deve sempre indurre il sospetto di un quadro sindromico particolare e andranno pertanto eseguite indagini volte a identificare eventuali anomalie congenite a carico di altri organi e apparati, che non sempre sono evidenti sul piano clinico.
Tra le malformazioni maggiori più frequentemente correlate a un quadro sindromico vanno ricordate le cardiopatie congenite (20%), le anomalie ano-rettali (45-50%), la labio-palatoschisi e la palatoschisi (rispettivamente 28-37% e 22-47%), l’ernia diaframmatica (20%) e i difetti in riduzione degli arti (40%). Alcune malformazioni maggiori poi, sebbene non correlabili esclusivamente a una specifica sindrome, sono riscontrate con maggiore frequenza in specifici quadri sindromici, basti pensare alla stenosi sopra-valvolare aortica nella sindrome di Williams, alla tetralogia di Fallot nella sindrome da microdelezione 22q11.2 e all’atresia esofagea nelle sindromi CHARGE, di Feingold e nell'associazione VACTER.
Malformazioni minori e note dismorfiche
Le malformazioni minori sono alterazioni morfo-strutturali di scarsa rilevanza clinica, che non alterano alcuna funzione e che, quindi, non richiedono alcun trattamento. Si parla di malformazione o anomalia minore quando questa è riscontrata in meno del 4% della popolazione di riferimento, per distinguerla dalla variante fenotipica, presente in più del 4% della popolazione.
L’esame dismorfologico rappresenta un momento fondamentale del processo diagnostico. Un corretto esame dismorfologico va eseguito in senso cranio-caudale, descrivendo eventuali anomalie minori riscontrate a carico di cranio, capelli, volto (fronte, sopracciglia, occhi, naso, filtro nasale, bocca, padiglioni auricolari, ecc), collo, torace, arti, mani/piedi, genitali esterni, cute. A tal proposito, è opportuno ricordare che difficilmente una singola anomalia minore è evocativa di un quadro sindromico preciso, ma è l’insieme delle caratteristiche fenotipiche del paziente a orientare verso una specifica sindrome.
Anomalie della crescita
La valutazione dei parametri auxologici e di eventuali anomalie della crescita è importante al fine di un corretto inquadramento diagnostico. Infatti, ogni anomalia dell’accrescimento staturo-ponderale (ritardo di crescita o iperaccrescimento) così come alterazioni della crescita della circonferenza cranica devono indurre nel pediatria il sospetto di una sindrome complessa. Andranno, quindi, sempre valutati peso, altezza/lunghezza, circonferenza cranica, lunghezza degli arti superiori e inferiori, larghezza delle braccia aperte (span) e rapporto tra span e altezza. Inoltre, sarà importante mettere in evidenza un’eventuale asimmetria corporea, dovuta ad emi-ipertrofia (come nel caso della sindrome di Beckwith-Wiedemann) o emi-ipotrofia (nella sindrome di Silver-Russell). È opportuno, però, ricordare che i parametri auxologici vanno sempre valutati in relazione alle caratteristiche familiari e al potenziale genetico del nostro paziente.
Per quanto riguarda il ritardo di crescita, questo può avere un esordio in epoca prenatale, manifestandosi con ritardo di crescita intra-uterino (IUGR) o rendersi evidente più tardivamente. Molti sono i quadri sindromici caratterizzati da IUGR: sindromi associate ad anomalie cromosomiche (sindrome di Down, trisomia 13, trisomia 18, sindrome di Turner, sindrome del Cri du Chat, ecc.), sindromi monogeniche (ad esempio sindrome di Cornelia de Lange, sindrome di Smith-Lemli-Opitz, sindrome di Aarskog, sindrome CHARGE), sindromi da anomalie dell’imprinting (sindrome di Silver-Russell) e displasie scheletriche (acondroplasia, ipocondroplasia, sindrome di Ellis-Van-Creveld, sindrome di Leri-Weill, ecc.). Alcuni quadri sindromici invece, tra cui ad esempio la sindrome di Noonan, sono caratterizzati da un ritardo di crescita che si manifesta nei primi mesi/anni di vita.
Anche un iperaccrescimento, sebbene meno frequente, deve far sospettare una sindrome malformativa complessa. Come visto per quanto riguarda il ritardo di crescita, anche l’iperaccrescimento può insorgere nella vita fetale, determinando un quadro di macrosomia neonatale, o manifestarsi dopo la nascita. Tra le sindromi con iperaccrescimento a evidenza neonatale, vanno ricordate la sindrome di Beckwith-Wiedemann, la sindrome di Sotos, la sindrome di Perlman e la sindrome di Bannayan-Riley-Ruvalcaba. Quadri caratterizzati da un iperaccrescimento che si manifesta più tardivamente sono, invece, la sindrome di Marfan e le sindromi marfanoidi. Queste sono caratterizzate da un’alta statura disarmonica, per cui sarà importante valutare lo span e il rapporto span/altezza.
Menzione particolare merita la sindrome di Prader-Willi: i pazienti affetti presentano ritardo di crescita intra-uterino e scarso accrescimento staturo-ponderale durante il primo anno di vita, cui fa seguito lo sviluppo di obesità secondaria ad iperfagia patologica. Altri quadri sindromici sono caratterizzati da obesità, ad esempio la sindrome di Bardet-Biedl e la sindrome di Cohen.
Infine, anche il riscontro di macrocefalia o microcefalia (circonferenza cranica rispettivamente > +2 DS e < -2 DS per età e sesso) deve fare sospettare una possibile causa sindromica, ma è fondamentale distinguere tra forme congenite ed acquisite.
Valutazione dello sviluppo psico-motorio e cognitivo
Anomalie dello sviluppo psico-motorio e ritardo mentale di vario grado sono frequentemente riconducibili a cause genetiche. Di conseguenza, di fronte ad un paziente con ritardo psico-motorio e/o disabilità intellettiva, è opportuno un approfondimento diagnostico volto ad escludere un eventuale quadro sindromico. In molti casi, il bambino può presentare una sindrome nota e facilmente riconoscibile (sindrome di Down, sindrome dell’X-fragile, sindrome di Angelman, ecc), ma oggi è sempre più frequente il riscontro di variazioni quantitative del DNA (micro-delezioni e micro-duplicazioni) in pazienti con disabilità intellettiva. L’introduzione degli array-GCH nello studio del DNA ha, infatti, consentito nell’ultimo decennio l’identificazione di anomalie criptiche e CNVs (Copy Number Variations) patologiche de novo e una sempre più accurata correlazione genotipo-fenotipo, contribuendo alla definizione di nuove sindromi.
È chiaro come la probabilità che un paziente con ritardo di sviluppo psico-motorio e/o ritardo mentale sia affetto da una sindrome complessa aumenta qualora coesistano note dismorfiche, malformazioni maggiori e/o anomalie dell’accrescimento.
Dal sospetto clinico alla diagnosi genetica
Dopo aver posto un sospetto clinico di sindrome complessa, è necessario procedere con indagini genetiche che lo confermino. In alcuni casi, il quadro clinico può essere evocativo per una sindrome nota: la diagnosi sarà pertanto “gestaltica” (“a colpo d’occhio”) e si procederà con test genetici mirati. Nel caso in cui, ad esempio, si sospetti una sindrome cromosomica (sindrome di Down, trisomia 13, trisomia 18, sindrome di Turner) si procederà con un esame del cariotipo. Si potrà, invece, ricorrere alla FISH (Fluorescence In Situ Hybridization) qualora il sospetto sia nei confronti, per esempio, della sindrome di Williams, ovvero all’analisi mutazionale del DNA se il quadro clinico è fortemente suggestivo per sindrome di Noonan (geni PTPN11, SOS1, ecc.). È bene però sapere che la maggior parte delle sindromi note mostra eterogeneità genetica, ovvero mutazioni di più geni responsabili di uno stesso fenotipo. Peraltro, ad oggi, di molti quadri sindromici non si conoscono tutte le alterazioni genetiche responsabili. È possibile quindi che un sospetto clinico, anche molto forte, non venga confermato da un corrispettivo genetico.
Molti pazienti, invece, non presentano un fenotipo evocativo per una sindrome nota, per cui non si potrà procedere con indagini mirate, ma sarà necessario un approccio diverso. Si potrà, in questi casi, ricorrere ad analisi mediante array-CGH, in grado di individuare alterazioni quantitative del DNA (micro-delezioni/micro-duplicazioni) mediante il confronto con un genoma di riferimento; oppure sarà possibile completare l’iter diagnostico con tecniche basate su NGS (Next Generation Sequencing), che consentono di sequenziare l’intero genoma o l’intero esoma o di studiare in maniera approfondita regioni d’interesse.
Al giorno d’oggi, il laboratorio di genetica ci fornisce innumerevoli strumenti, ciascuno con la sua caratteristica: tecniche di citogenetica classica (cariotipo standard e cariotipo ad alta risoluzione), tecniche di citogenetica molecolare (FISH), genetica molecolare (sequenziamento genico, analisi mutazionale del DNA), test biochimici, array-CGH, studio dei riarrangiamenti subtelomerici, NGS. Nonostante ciò, una diagnosi eziologica viene raggiunta nel 70-75% dei casi, mentre nel restante 25-30% dei pazienti non si giunge ad una diagnosi certa. Per questi pazienti sarà necessario un follow-up periodico: si potrà infatti avere, nel tempo, un’evoluzione fenotipica del paziente o la comparsa di nuovi segni clinici ad insorgenza età-dipendente, ovvero la possibilità di ulteriori approfondimenti con la definizione di nuove tecnologie.
Bibliografia
- Special Issue: Elements of Morphology: Standard Terminology. Am J Med Genet 2009, 149 A: 1-127.
- Selicorni A, Zampino G, Memo L, Scarano G. Le sindromi malformative: una guida per il pediatra. Pacini Editore 2014.
- Corsello G, Giuffrè M, Piccione M. Il neonato con anomalie congenite multiple: inquadramento e nosologia. Prospettive in Pediatria 2013: 149-57.
- Piro E, Consiglio V, Agrifoglio M, et al. Diagnosis and follow-up of complex congenital malformations/mental retardation (MRA/MR). Acta Medica Mediterranea 2013, 29: 321-5.
- Weise A, Mrasek K, Klein E, et al. Microdeletion and microduplication syndromes. J Histochem Cytochem 2012, 60: 346-58.
Sindrome di Alström
Elisa Parolo, Francesca Dassie, Francesca Favaretto, Gabriella Milan, Pietro Maffei
Azienda Ospedaliera di Padova, Clinica Medica 3^, DIMED
(aggiornato al 21 luglio 2016)
Epidemiologia e genetica
La sindrome di Alström (ALMS) (OMIM 203800) è una malattia monogenica molto rara, descritta per la prima volta nel 1959 da CH Alström, a ereditarietà mendeliana autosomica recessiva. La prevalenza è di circa 1:1.000.000 nella popolazione generale e sono stati descritti finora circa 1000 casi in tutto il mondo.
È una malattia sistemica a espressione fenotipica variabile, la cui caratteristica principale è una fibrosi generalizzata a eziologia ancora sconosciuta, che porta a una progressiva insufficienza multi-organo. I pazienti hanno un’aspettativa di vita ridotta, che raramente supera i 50 anni di età. Vi è una grande variabilità nella clinica, anche nei pazienti portatori della stessa mutazione, sia per quanto riguarda la gravità dei sintomi, sia per l’età di esordio, che può avvenire alla nascita, in età infantile o durante l’adolescenza.
L’ALMS è causata da mutazioni nel gene ALMS1, localizzato sul braccio corto del cromosoma 2 (2p13); ALMS1 contiene 23 esoni, che codificano una proteina di 4.169 aminoacidi, la cui funzione è solo in parte conosciuta. Ad oggi sono state individuate più di 200 mutazioni, la maggior parte nonsense, delezioni ed inserzioni, raggruppate negli esoni 8, 10 e 16. La proteina prodotta da ALMS1 normalmente è localizzata nei corpi basali, in prossimità del cilio e nei centrosomi, ed è quindi implicata sia nel funzionamento delle cilia sia nel trasporto citoplasmatico del sistema micro-tubulare.
L’ALMS può essere classificata nel gruppo di malattie definite come ciliopatie, che hanno in comune una disfunzione ciliare, a cui appartiene anche la sindrome di Bardet-Biedl, con la quale ALMS condivide i difetti molecolari a livello dei corpi basali e alcune caratteristiche cliniche.
Caratteristiche cliniche
Tratti somatici: sono caratteristici, con occhi infossati, viso arrotondato, iperostosi frontale, orecchie piccole e spesse, stempiatura prematura fino a quadri di calvizie con capelli sottili; inoltre, è spesso presente brachidattilia con dita tozze, e piedi piatti. Frequente è anche la presenza di scoliosi o cifosi, e anomalie dentarie. Queste caratteristiche somatiche si rendono più evidenti con la crescita del paziente.
Sviluppo mentale: la maggior parte dei pazienti presenta un’intelligenza nella norma, soprattutto se la diagnosi della malattia è precoce e vengono forniti quindi tutti gli strumenti per un normale raggiungimento delle tappe fondamentali dello sviluppo. Sono stati riportati rari casi di ritardo mentale, che potrebbero però essere legati essenzialmente a supporto educazionale scarso o assente. Inoltre, si sono visti casi che presentano problemi neurologici, come episodi di assenza, epilessia, comportamento autistico.
Deficit visivi: già poche settimane dopo la nascita si verifica una disfunzione retinica, con insorgenza di nistagmo e fotofobia; all’elettroretinogramma si evidenzia una grave compromissione della retina, caratterizzata da distrofia dei coni e attività ridotta dei bastoncelli. I potenziali visivi evocati risultano molto ridotti. Con il tempo vi è una progressiva degenerazione retinica, che si estrinseca in un quadro di retinite pigmentosa, con perdita progressiva di funzionalità fino alla cecità, che si instaura generalmente entro la seconda decade di vita. Anche se il processo è irreversibile, si può intervenire precocemente con supporto sia medico che educazionale (ausili visivi e informatici dedicati, apprendimento della lettura Braille).
Deficit uditivi: nella maggior parte dei casi c’è una progressiva sordità di tipo neuro-sensoriale bilaterale già nella prima decade di vita, che evolve solitamente verso forme moderate-severe. Il processo può essere favorito da otiti acute ricorrenti, che compromettono la via trasmissiva. Anche se solitamente il deficit si instaura dopo l’acquisizione del linguaggio, l’utilizzo di adeguate protesi acustiche facilita un corretto sviluppo psico-motorio. Nei casi più gravi si è dimostrata efficace la terapia chirurgica con impianti cocleari.
Alterazioni metaboliche: l’obesità è una caratteristica clinica costante, che si manifesta precocemente. Peso e lunghezza sono normali alla nascita, ma già nei primi mesi di vita vi è un progressivo incremento ponderale, con frequente concomitante iperfagia; è verosimile che ciò accada per una disfunzione centrale dei centri regolatori del'appetito. Con l’avanzare dell’età, si assiste talvolta a una progressiva normalizzazione del peso; questo peculiare quadro clinico sottende meccanismi fisio-patologici tuttora poco conosciuti. È stato riportato un marcato aumento del tessuto adiposo sotto-cutaneo addominale e la presenza di lipodistrofia soprattutto negli adulti. Spesso si riscontra ipertrigliceridemia, non sempre accompagnata da ipercolesterolemia; i livelli di trigliceridi possono essere tali da causare in alcuni casi pancreatiti. È possibile intervenire sui problemi ponderali e metabolici con l’introduzione di una dieta personalizzata e incentivando l’attività fisica, quest’ultima programmata tenendo conto del deficit visivo. Si riscontra frequentemente marcata insulino-resistenza, iperinsulinemia e ridotta tolleranza al glucosio, talvolta già nel primo anno di vita, che porta spesso allo sviluppo di franco diabete mellito tipo 2 prima dei vent’anni. Molto spesso questa alterazione del metabolismo glucidico è accompagnata dall’insorgenza di acanthosis nigricans. Anche in questo caso una dieta povera di carboidrati, e, nei casi meno controllati, l’impiego di anti-diabetici orali (metformina, incretine), mirano a ottenere un compenso glicemico adeguato, allo scopo di evitare l’insorgenza di complicanze. La marcata insulino-resistenza rende la terapia con insulina poco efficace anche con dosaggi elevati.
Alterazioni endocrine: esiste generalmente ipogonadismo iper- o ipogonadotropo, con frequenza maggiore nei maschi rispetto alle femmine. Nei maschi i livelli di testosterone sono bassi, vi è un ridotto sviluppo degli organi riproduttivi e i caratteri sessuali secondari tendono a essere normali; talvolta compare ginecomastia. Nelle femmine l’ipogonadismo si può rendere manifesto alla pubertà, quando vi è un ritardo nella comparsa del menarca e dei caratteri sessuali secondari; le pazienti possono presentare un quadro di iperandrogenismo, irsutismo e alopecia. Le ovaie possono avere formazioni cistiche e fibrosi, con follicoli primari o secondari in numero esiguo o addirittura assenti; i cicli mestruali molto spesso sono irregolari con oligo-amenorrea. Oltre alla clinica, è quindi importante determinare e monitorare i livelli di gonadotropine e degli ormoni sessuali all’epoca della pubertà, per un’eventuale terapia sostitutiva nei maschi e l’utilizzo di estroprogestinici o insulino-sensibilizzanti nelle femmine. I pazienti ALMS hanno caratteristicamente una bassa statura ed è stata descritta una maggiore prevalenza di sella vuota; prima della pubertà, vi è una crescita staturale oltre il cinquantesimo percentile, con un’età ossea più avanzata di circa due-tre anni. Tuttavia, alla pubertà si può osservare un rapido declino della velocità di crescita, e questo comporta una statura finale inferiore alla norma. È stato studiato l’asse GH-IGF1, riscontrando in molti casi una ridotta secrezione di GH in risposta ai test di stimolo massimali. La terapia sostitutiva con GH rimane controversa, nonostante si sia visto un potenziale beneficio su composizione corporea, metabolismo glucidico e funzionalità cardiaca. Altro disordine endocrino frequente è l’ipotiroidismo di tipo centrale, mentre in alcuni pazienti insorge ipotiroidismo di tipo primario, a volte subclinico.
Alterazioni cardiache: nella maggior parte dei casi si ha cardiomiopatia dilatativa, con conseguente insufficienza cardiaca, che molto spesso insorge improvvisamente già nei primi mesi di vita. In seguito a trattamento è stato descritto un apparente recupero della funzionalità cardiaca. Tuttavia, può recidivare durante l’adolescenza o in età adulta, con coinvolgimento di entrambi i ventricoli e rapida progressione dell’insufficienza cardiaca, che porta a prognosi severa; infatti, la cardiomiopatia dilatativa rappresenta una delle cause più importanti di mortalità in questi pazienti. In una minoranza di soggetti, l’insufficienza cardiaca compare per la prima volta nell’adolescenza o in età adulta, verosimilmente in seguito a un lento processo di fibrosi, che coinvolge il muscolo cardiaco. La forma neonatale e quella dell’adulto sembrano avere meccanismi fisio-patologici diversi. In rari casi è stato effettuato trapianto di cuore o cuore-polmoni con risultati discordanti.
Alterazioni epatiche: nei pazienti con ALMS la disfunzione epatica mostra rilevante variabilità fenotipica, sia per quanto riguarda l’età di insorgenza, sia per decorso e prognosi; spesso esordisce clinicamente già nell’infanzia, con aumento delle transaminasi e steatosi. La steatosi epatica si associa frequentemente a obesità e diabete mellito; un meccanismo favorente l’accumulo di grasso nel fegato potrebbe essere legato proprio alla marcata insulino-resistenza. Questo quadro può peggiorare, portando a steatosi di grado moderato-severo, fino all’insorgenza di cirrosi epatica complicata. La rottura delle varici esofagee è una temibile complicanza dell’ipertensione portale osservata in alcuni casi.
Alterazioni renali: anche per questo aspetto, vi è un’ampia variabilità di presentazione clinica, in quanto l’età di esordio, la gravità della malattia e la relativa velocità di progressione sono diverse da soggetto a soggetto. Inoltre, vi sono casi che presentano solo lieve insufficienza renale cronica, altri in cui vi è albuminuria e rapida progressione fino a insufficienza renale terminale. Alcuni pazienti manifestano anche problemi urologici, con disfunzione dello sfintere vescicale, tali da richiedere, in alcuni casi, la cateterizzazione transitoria o permanente. In alcuni pazienti è stato effettuato con successo il trapianto di rene.
Alterazioni respiratorie: fin dall’infanzia si manifestano infezioni ricorrenti, che possono complicarsi con bronchite, sinusite cronica o asma; il quadro clinico può portare nell’adulto a bronchiectasie o bronco-pneumopatia cronica ostruttiva. È comune anche il riscontro di ipertensione polmonare. In certi casi è stata documentata la presenza di grave fibrosi interstiziale del parenchima polmonare.
Approccio diagnostico e terapeutico
Sono stati elaborati criteri diagnostici clinici, distinti tra maggiori e minori, che permettono di formulare la diagnosi nelle diverse fasce di età, anche quando non viene individuata una mutazione di ALMS1 in entrambi gli alleli in omozigosi (gold standard).
| Criteri diagnostici per ALMS | ||
| Maggiori (validi per tutte le fasce di età) |
|
|
| Minori | < 2 anni |
|
| 3-14 anni |
|
|
| > 15 anni |
|
|
La diagnosi è comprovata da:
- nel paziente < 2 anni: 2 criteri maggiori o 1 maggiore + 2 minori;
- nella fascia 3-14 anni: 2 criteri maggiori oppure 1 criterio maggiore + 3 minori;
- nella fascia > 15 anni: 2 criteri maggiori + 2 minori oppure 1 criterio maggiore + 4 minori.
ALMS si manifesta inizialmente con disturbi visivi, può essere confusa con altre retinopatie, come ad esempio l’amaurosi congenita di Leber. Inoltre, poichè l’esordio può avvenire con cardiomiopatia e insufficienza cardiaca, questo quadro potrebbe essere erroneamente classificato come cardiomiopatia dilatativa infantile idiopatica o miocardite. Pertanto, è fondamentale una diagnosi differenziale corretta e supportata dall’indagine genetica.
Purtroppo non è ancora disponibile una terapia mirata per i pazienti ALMS, da cui l’importanza di una diagnosi precoce per un corretto approccio multi-disciplinare, al fine di prevenire le complicanze e migliorare sia la qualità che l’aspettativa di vita. Nella prima infanzia, è di fondamentale importanza sopperire ai deficit di tipo sensoriale, per un corretto sviluppo psico-motorio; inoltre devono essere approntate adeguate strategie per il trattamento dell’obesità e dell’iperfagia. Alterazioni metaboliche ed eventuali disfunzioni endocrine possono essere gestite con appropriata terapia medica. Per i problemi clinici maggiori, come la cardiopatia o le alterazioni epatiche e renali, è di fondamentale importanza coordinare l’intervento dei diversi specialisti, per ottimizzare la funzionalità dei vari organi e migliorare l’outcome a lungo termine.
Bibliografia
- Marshall JD, Muller J, Collin GB, et al. Alström syndrome: mutation spectrum of ALMS1. Hum Mutat 2015, 36: 660-8.
- Marshall JD, Maffei P, Collin GB, Naggert JK. Alström syndrome: genetics and clinical overview. Curr Genomics 2011, 12: 225-35.
- Marshall JD, Beck S, Maffei P, Naggert JK. Alström Syndrome. Eur J Hum Genet 2007, 15: 1193-202.
- Marshall JD, Bronson RT, Collin GB, et al. New Alström syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med 2005, 165: 675–83.
Siti web
- Alström Syndrome International
- Alström Syndrome UK
- ASSAI Onlus – Associazione Sindrome di Alström Italia
Sindrome di Down
Giovanni Corsello & Vincenzo Antona
Dipartimento di Scienze per la Promozione della Salute e Materno Infantile "G. D'Alessandro", Università degli Studi di Palermo
La sindrome di Down (SD) o trisomia 21 è caratterizzata da un deficit della crescita ed intellettivo di grado variabile, da peculiari anomalie minori del volto e da malformazioni maggiori a carico di diversi organi e apparati, in particolare cardiopatie congenite, anomalie gastro-intestinali e genito-urinarie.
EPIDEMIOLOGIA
La SD è l'anomalia cromosomica più frequente. I dati epidemiologici sono sostanzialmente sovrapponibili in tutto il mondo: la prevalenza nei nati vivi è di 1:700-1000; la prevalenza nella popolazione generale è di 1:2000.
Oggi, grazie alle cure più attente che vengono offerte, la vita media dei soggetti con SD è di 60 anni (1). Le principali cause di morte in età adulta sono le malattie respiratorie (20-40%) e circolatorie (25-40%), incluse le conseguenze di una patologia cardiaca congenita. Lo sviluppo di demenza diventa considerevole dopo i 40 anni, contribuendo a circa un terzo delle morti.
EZIOPATOGENESI
Le caratteristiche cliniche dei soggetti con SD sono causate dalla presenza di un extra-cromosoma 21 che può essere il risultato di diversi eventi.
- Trisomia completa (90-95% pazienti con SD): causata da un evento di non disgiunzione, cioè dalla mancata separazione dei cromosomi della coppia 21 durante la meiosi, per cui in uno dei due genitori la cellula germinale, cellula uovo o spermatozoo, contiene una doppia copia di cromosomi 21 invece che uno solo. La fecondazione di questa cellula germinale porta alla formazione di uno zigote trisomico per il cromosoma 21. Questa trisomia si verifica spontaneamente in modo imprevedibile e non dipende da altre anomalie cromosomiche (trisomia libera o primaria). Nel 90% dei casi la non-disgiunzione è di origine materna. La trisomia completa è l'evento più frequente di aneuploidia nella specie umana.
- Mosaicismo cromosomico (3%): origina solitamente da non-disgiunzione mitotica di uno zigote inizialmente a cariotipo normale. Nello stesso individuo, quindi, sono presenti 2 linee cellulari, una con normale assetto cromosomico e l'altra con trisomia del cromosoma 21. Il mosaicismo sembra essere correlato a un quadro clinico più attenuato, anche se esiste un'ampia variabilità dipendente dalla percentuale di cellule trisomiche, diversa nei differenti tessuti e organi dello stesso individuo.
- Traslocazione sbilanciata (4%): che coinvolge un cromosoma 21 e i cromosomi 13, 14, 15 o 22 (traslocazione robertsoniana) o il cromosoma 21 stesso. Questo assetto può insorgere casualmente nell'individuo affetto o può derivare dalla segregazione sbilanciata di una traslocazione bilanciata in uno dei due genitori (trisomia 21 secondaria). Se il bimbo presenta una traslocazione, va sempre esclusa nei genitori la presenza di una traslocazione bilanciata.
- Trisomia parziale: coinvolgente solo la parte distale del braccio lungo del cromosoma 21 coinvolta in un riarrangiamento strutturale, quale una traslocazione sbilanciata o una duplicazione parziale. Lo studio genetico e fenotipico di questi pazienti ha permesso di individuare una regione critica per la definizione del fenotipo clinico associato alla SD a livello della banda 21q22.
MANIFESTAZIONI CLINICHE
Tratti del volto peculiari: viso tondo con brachicefalia, plica nucale, rime palpebrali oblique, epicanto, naso piccolo con sella nasale piatta, padiglioni auricolari piccoli e a impianto basso, palato ogivale, macroglossia e micrognatia.
Altre anomalie somatiche: collo corto, capelli sottili, solco palmare unico, clinodattilia del 5° dito della mano, sindattilia.
Ipotono
Rallentamento della crescita staturale: si verifica soprattutto tra i 9 e i 24 mesi e tra i 10 e i 17 anni anche in assenza di patologie malformative maggiori associate. La statura media è pari a circa 153 cm nei maschi e 143 cm nelle femmine. La bassa statura è dovuta alla brevità degli arti, mentre il tronco è di dimensioni pressochè normali.
Rallentamento della crescita ponderale: è caratteristico soprattutto dei primi 2 anni di vita ed è associato alle cardiopatie congenite e alle problematiche alimentari (RGE, macroglossia, ipotono).
Ritardo psico-motorio: ritardo nel raggiungimento delle principali tappe dello sviluppo motorio a causa dell'ipotono e ritardo mentale di grado variabile, con importante interessamento dell'area del linguaggio.
Malformazioni cardio-vascolari: difetti del setto inter-ventricolare (DIV), difetto inter-atriale (DIA) tipo ostium II, tetralogia di Fallot, pervietà del dotto di Botallo e canale atrio-ventricolare comune. I difetti cardiaci congeniti sono presenti nel 50-60% dei bambini con SD. Alcuni di questi difetti si risolvono spontaneamente (Botallo pervio, spesso DIA, a volte DIV), gli altri comportano un intervento cardio-chirurgico nei primi mesi/anni di vita, da valutare a seconda della compromissione clinica e cardiologica e dell'accrescimento ponderale (2).
Altre malformazioni:
- criptorchidismo = 14% (contro 0.8% della popolazione generale);
- atresia gastro-duodenale = 8-12%, più spesso a carico del duodeno, a volte esofagea; richiede un intervento chirurgico tempestivo;
- morbo di Hirschprung = 2-4%;
- pancreas anulare (< 1%).
DIAGNOSI
La diagnosi di SD viene solitamente sospettata alla nascita sulla base delle anomalie minori del volto e delle eventuali anomalie maggiori associate, che nel loro insieme caratterizzano il fenotipo tipico di questa sindrome. La diagnosi clinica viene confermata con certezza solo dall'indagine citogenetica eseguibile sui linfociti periferici.
DIAGNOSI PRENATALE
Le principali indicazioni all'indagine citogenetica prenatale sono:
- età materna avanzata (≥ 35 anni);
- genitori con precedente figlio affetto da patologia cromosomica come la SD;
- genitore portatore di riarrangiamento strutturale non associato ad effetto fenotipico;
- genitori con aneuploidie dei cromosomi X o Y compatibili con la fertilità;
- malformazioni fetali evidenziate mediante l'ecografia;
- probabilità ≥ 1:250 che il feto sia affetto da SD (o da alcune altre aneuploidie) sulla base dei parametri di screening ecografici o biochimici (TN, bi-test, tri-test ecc.).
In caso di indicazione all'indagine citogenetica, viene offerta la possibilità di eseguire la villocentesi o l'amniocentesi, procedure invasive e quindi correlate ad aumentato rischio di abortività, ma che garantiscono la certezza diagnostica.
COMPLICANZE MEDICHE
Nella SD alcune problematiche cliniche sono più frequenti e compaiono prima rispetto alla popolazione generale. In particolare le seguenti richiedono un'attenta valutazione.
Endocrinopatie (3):
- ipotiroidismo: presente nel 15-37% dei pazienti in tutte le età;
- ipertiroidismo: leggermente più comune (0.65%) rispetto alla popolazione generale;
- sovrappeso e obesità: a causa del metabolismo basale ridotto, sedentarietà ed eccessi alimentari;
- osteoporosi: la SD è un fattore di rischio indipendente per osteoporosi; la prevalenza di fratture è del 55% per le ossa lunghe e del 30% per i corpi vertebrali sopra i 50 anni;
- diabete mellito: si sviluppa in circa l’1-14% dei bambini e degli adolescenti.
Patologie cardio-vascolari:
- prolasso della mitrale (46%) e/o insufficienza aortica (17%): possono svilupparsi anche negli adolescenti e negli adulti senza cardiopatie già note;
- ipertensione polmonare.
Patologie gastro-intestinali:
- reflusso gastro-esofageo;
- disfagia: il 50% dei pazienti con difficoltà nella deglutizione ha rischio di aspirazione;
- celiachia: può svilupparsi a tutte le età, con prevalenza complessiva del 7-17%. Può essere asintomatica o presentarsi con sintomi aspecifici, quali cambiamenti del comportamento o dell'umore, così come con perdita di peso e diarrea.
Malattie ematologiche-oncologiche:
- policitemia nei neonati: soprattutto per compenso in presenza di cardiopatia congenita;
- malattia mielo-proliferativa transitoria alla nascita (TMD): è una forma di leucemia auto-limitantesi, che per ragioni sconosciute regredisce spontaneamente nell'arco di 2-3 mesi dalla nascita. Si manifesta quasi esclusivamente nei neonati con SD (prevalenza del 10% nella popolazione Down);
- mielodisplasia, leucemia mieloide acuta e leucemia linfoblastica acuta: il rischio di leucemia è 10-20 volte maggiore rispetto alla popolazione generale, ma spesso risulta inusualmente sensibile alla chemioterapia. Il rischio oncologico diminuisce all'avanzare dell’età, con più del 90% dei casi prima dei vent'anni. I pazienti con SD, inoltre, hanno molto frequentemente leucopenia, macrocitosi idiopatica, lieve policitemia, spesso senza una patologia sottostante. Occorre comunque mantenere un alto indice di sospetto per leucemia, anche in età adulta.
Malattie polmonari:
- la suscettibilità alle infezioni respiratorie é elevata: la polmonite è la prima causa di ricovero e la seconda causa di morte dopo le cardiopatie congenite. Il tasso di mortalità legata alla polmonite aumenta proporzionalmente con l'età;
- apnee ostruttive del sonno (50-70%): la patogenesi è legata a diversi fattori, quali obesità, macroglossia, ipertrofia adeno-tonsillare, ricorrenti infezioni delle alte vie aeree, ipoplasia ossea del terzo medio-inferiore del volto, reflusso gastro-esofageo, ipotono faringeo, iperlassità faringea, stenosi sotto-glottica. Possono comparire a tutte le età e presentarsi con cambiamenti dell'umore o del comportamento, declino delle competenze, fatica, sonnolenza diurna, episodi di dispnea notturna;
- frequenti sono gli accessi ospedalieri per bronchioliti che necessitano di ventilazione assistita.
Difetti uditivi: ipoacusia (75%) di tipo neuro-sensoriale o conduttivo (a causa di aumentata suscettibilità a otite media acuta e cronica).
Malattie oculari (60%): cataratta congenita, glaucoma, difetti di rifrazione, strabismo, nistagmo, cheratocono (15% adolescenti), cataratta senile (13%), aree di degenerazione retinica.
N.B. I difetti neuro-sensoriali possono essere interpretati come decadimento delle capacità intellettive e cognitive. Sono situazioni che vanno ricercate (in molti casi il paziente difficilmente segnalerà il disturbo perchè non in grado di percepirlo o esprimerlo compiutamente) e discriminate, poichè la correzione di un difetto di vista e udito può permettere alla persona che ne è affetta di tornare a inserirsi rapidamente nel suo ambiente sociale.
Patologie neurologiche (4,5):
- epilessia (5-8%): può comparire a tutte le età; nella prima infanzia anche sotto forma di spasmi infantili. Le crisi possono comportare declino cognitivo ulteriore se non controllate;
- compressione spinale cervicale: secondaria ad instabilità atlanto-assiale nei bambini e a patologia degenerativa della colonna vertebrale negli adulti;
- demenza di Alzheimer: si sviluppa a partire dalla 5° decade, con una prevalenza del 50-70% dopo i 60 anni. Esistono differenze della presentazione clinica tra le persone con e senza SD. Segni e sintomi sono rappresentati più spesso da crisi epilettiche (58%), segni neurologici focali (46%), aprassia del cammino, incontinenza, cambiamenti nella personalità (46%, in particolare minor capacità di adattamento), apatia (36%). Gli strumenti diagnostici tradizionali sono inaffidabili e impraticabili in circa la metà dei pazienti; nonostante l'esistenza di diverse opzioni alternative, non c'è consenso su quale sia il metodo migliore di valutazione.
Patologie psichiatriche (6,7):
- depressione: comunemente si presenta con ritiro sociale, diminuzione dell'appetito, riduzione dell'eloquio, ma può esordire anche con declino delle abilità e incontinenza urinaria ed essere scambiata per demenza di Alzheimer. Può rappresentare essa stessa un segno precoce di demenza. Spesso è responsiva alla terapia medica;
- ansietà;
- tratti ossessivo-compulsivi;
- problemi comportamentali;
- disturbo dello spettro autistico: 10 volte più frequente rispetto alla popolazione generale;
- regressione: rara, si verifica in adolescenza e comporta perdita rapida ed atipica delle abilità precedentemente acquisite, delle capacità di socializzazione, delle attività quotidiane, con un incremento dei comportamenti maladattativi;
- psicosi.
Patologie ortopediche: gran parte della patologia ortopedica è secondaria alla lassità dei legamenti tipica della SD:
- instabilità atlanto-assiale: ha una frequenza di circa il 10-15% ma dà sintomi nell'1% dei pazienti con SD. I sintomi neurologici più frequentemente evidenti sono stanchezza nel mantenere a lungo la stazione eretta, deambulazione incerta e barcollante associata a frequenti cadute, paraparesi, torcicollo persistente, posizioni viziate del capo, incontinenza urinaria, iper-reflessia, clono. Questi segni possono rimanere relativamente stabili per mesi o anni, mentre talora progrediscono. Esordio comune dopo i 12 anni (5);
- lussazione congenita dell'anca, ginocchio valgo, piede piatto lasso, lussazione rotulea, metatarso varo, alluce valgo e scoliosi evolutiva;
- artrite e artrosi.
Manifestazioni dermatologiche (80%): follicoliti (60%), cheratosi palmo-plantare, cheilite angolare e sulle labbra, xerosi cutanea, disidrosi, dermatite seborroica, lingua fissurata, cutis marmorata, micosi, vitiligo e alopecia.
Problematiche odonto-stomatologiche (9): ritardo di eruzione cutanea, malocclusione, disturbi della masticazione, gengiviti, periodontopatie. Presenti nel 60-100% dei casi sopra i 30 anni.
Altre patologie (più rare ma descritte in una percentuale superiore rispetto alla popolazione generale): artrite reumatoide giovanile (< 1%), epatite cronica attiva e sindrome poliendocrina autoimmune tipo I.
Condizioni meno frequenti: gli individui con SD presentano un rischio più basso rispetto alla popolazione generale per:
- tumori solidi come le neoplasie cervicali, mammarie, polmonari e prostatiche;
- patologia coronarica;
- ipertensione.
SESSUALITÀ E FERTILITÀ
La maturità sessuale nelle ragazze con SD si verifica regolarmente per quanto riguarda il ciclo mestruale, anche se talora un po' ritardata in termini di età di comparsa; il flusso mestruale ha di solito una durata fisiologica. Molti cicli mestruali sono anovulatori, ma le donne Down possono essere fertili. La menopausa compare precocemente (26-40 anni).
Nei maschi viene segnalata infertilità per ipo- o azoospermia.
Nella pubertà i ragazzi e le ragazze con SD hanno le stesse emozioni, pulsioni e desideri sessuali dei loro coetanei, in particolare ricercano il legame affettivo con l'altro sesso.
FOLLOW-UP (10,11)
La gestione medica insieme al contesto familiare, gli interventi precoci e la formazione professionale possono incidere profondamente sul livello di funzionamento dei bambini e degli adolescenti con SD e facilitare il passaggio all'età adulta.
Il follow-up si organizza attorno alle problematiche specifiche delle varie fasce di età. Alcune aree invece richiedono una valutazione continua e dovrebbero essere riesaminate ad ogni bilancio di salute.
| Tempistiche per il follow-up | ||
| Tutta la vita a cadenza annuale | Monitoraggio di peso, altezza e curva di crescita, facendo riferimento alle curve standardizzate per la popolazione generale. Nutrizione e attività fisica per mantenere il giusto peso: la prevenzione dell'obesità deve iniziare precocemente e comprendere scelte alimentari, interventi comportamentali, attività fisica e vita sociale attiva (lavoro, sport, tempo libero). La dieta dovrebbe essere programmata in modo tale da privilegiare cibi con alto contenuto di fibre e poveri di grassi e calorie. L'apporto calorico totale dovrebbe essere inferiore a quello giornaliero raccomandato per la popolazione generale. In questo stile di vita va tenuta in considerazione la necessità di evitare l'instaurarsi di una stipsi ostinata, causa di disagi psicologici spesso non espressi dall'individuo. Supporto a disposizione della famiglia. Impatto della patologia sulla famiglia e sull'adattamento degli altri figli. Programmi di supporto finanziario e medico per i quali il bambino e la famiglia sono candidabili. Prevenzione di danni e abusi. Discussione sulle terapie considerate complementari e alternative. |
|
| Dopo la diagnosi prenatale | Consigliare gli esami che aiutino a definire la prognosi (es. l'ecocardiogramma fetale identifica il 15-75% delle malformazioni cardiache congenite) e inviare da subito a valutazione specialistica in caso di reperti patologici. Discutere di continuazione/interruzione di gravidanza, allevamento del bimbo in famiglia, affidamento, adozione. Se la gravidanza continua, pianificare il parto e le cure neonatali con la famiglia e il ginecologo; offrire contatti e informazioni sulle associazioni locali, suggerire una valutazione genetica per una discussione più approfondita sugli esiti clinici, la loro variabilità, il tasso di ricorrenza, le opzioni riproduttive future, la valutazione del rischio di altri familiari. |
|
| Dalla nascita al I mese |
Il primo passo è riesaminare la storia familiare, le informazioni prenatali, in modo particolare se sia già stato fatto uno studio cromosomico o una consulenza genetica. Un altro figlio con SD o differenze nello sviluppo o aborti potrebbero essere indizi significativi della presenza in famiglia di una traslocazione.
|
|
| Da 1 mese ad 1 anno | Riesaminare il rischio di otite media. Se il bimbo ha superato lo screening, ripetere i potenziali evocati a 6 mesi per conferma. Se la membrana timpanica non è ben visualizzabile, effettuare un timpanogramma e far esaminare l'orecchio da un otorinolaringoiatra ogni 3-6 mesi finchè il timpano non sia ben visibile e il timpanogramma affidabile. Discutere con i genitori dei sintomi di apnea notturna. Entro i primi 6 mesi, consigliare una valutazione oculistica per strabismo, cataratta, nistagmo. Controllare la visione a ogni visita. Ripetere i dosaggi di TSH e FT4 a 6 e 12 mesi poi annualmente. Monitorare i bimbi con difetti cardiaci per sintomi riconducibili a insufficienza, compresi tachipnea, difficoltà nutrizionali, rallentamento di crescita. In caso di difetti inter-ventricolari ampi o ostruzione polmonare, l'intervento dovrebbe essere eseguito entro i 4 mesi di vita per limitare il rischio di ipertensione polmonare. Dosare la concentrazione di emoglobina, ferritina e PCR a un anno poi annualmente. Somministrare i vaccini raccomandati e quello per l'influenza. |
|
| Da 1 a 5 anni | Informare genitori su sintomi riferibili a instabilità atlanto-assiale e sulle attività e gli sport più a rischio per danno alla colonna vertebrale. Approfondire con radiografia cervicale solo i casi sintomatici; visita ortopedica dopo circa 6-8 mesi dalla deambulazione autonoma. Screening per celiachia solo nei casi sintomatici. A partire dai 4 anni, consigliare una poli-sonnografia anche negli asintomatici (poca correlazione tra quello che riportano i genitori e i risultati della poli-sonnografia). Discutere dell'obesità come fattore di rischio per apnee notturne. Mantenere il follow-up cardiologico per i bambini con lesioni cardiache anche dopo la loro completa riparazione, per il rischio di danno residuo/ricorrente e di ipertensione polmonare. In tutte le persone che hanno avuto alla nascita una cardiopatia congenita correttasi spontaneamente o con intervento chirurgico, è sempre indispensabile la profilassi per l'endocardite batterica in caso di manovre chirurgiche invasive, comprese quelle odontoiatriche. Ricercare sintomi neurologici e critici. Valutare gli interventi precoci come terapia fisica ed occupazionale e logopedia. Somministrare il vaccino anti-pneumococcico polivalente. Informare i genitori che il ritardo di eruzione dentale e l'ipodontia sono comuni. Discutere almeno una volta delle eventuali gravidanze future, del rischio di ricorrenza e della disponibilità della diagnosi prenatale. |
|
| Ogni 6 mesi | Riesaminare il rischio di ipoacusia legata ad otite media fino quando i livelli uditivi sono stabilizzati bilateralmente (in genere attorno ai 4 anni, poi annualmente). | |
| Ogni anno | Valutazione oftalmologica. Visita odontoiatrica. Dosaggio di TSH e FT4. Dosare Hb, ferritina, PCR. Somministrare il vaccino anti-influenzale. |
|
| Dai 5 ai 13 anni | Monitoraggio cardiaco individualizzato. Ricercare sintomi riconducibili a malattia celiaca. Ricercare sintomi neurologici. Ricercare sintomi di apnee notturne. Ricercare sintomi dermatologici. Discutere degli interventi e degli indirizzi scolastici appropriati, facilitando le fasi di passaggio. Discutere delle abilità, dell'autonomia e dello sviluppo del senso di responsabilità del paziente. Incoraggiare le autonomie. Valutare le problematiche comportamentali, escludere una causa organica e consigliare eventuale consulenza specialistica. Discutere dei cambiamenti legati alla pubertà, della fertilità e della contraccezione. Consigliare una valutazione ginecologica con eco pelvica per le ragazze adolescenti. |
|
| Ogni anno | Valutazione audiologica. Visita ortopedica. Visita odontoiatrica. Dosaggio di TSH e FT4. Dosaggio di Hb, ferritina e PCR. |
|
| Ogni 2 anni | Valutazione oculistica | |
| Dai 13 ai 18 anni | Ricerca sintomi di malattia celiaca. Monitoraggio cardiologico individualizzato. Monitoraggio obesità e sintomi di apnea notturna. Ricerca segni neurologici. Discutere dei problemi relativi al passaggio all'età adulta: invecchiamento precoce, rischio di demenza di Alzheimer, necessità di istituire un tutore, piani finanziari a lungo termine. Discutere di un appropriato indirizzo scolastico e della transizione scuola-lavoro. Discutere con il paziente e la famiglia del rischio di ricorrenza di SD nel caso in cui la ragazza affronti una gravidanza. Continuare a incoraggiare le autonomie; fornire un'educazione sessuale accessibile al paziente. |
|
| Ogni anno | Dosaggio TSH e FT4 Dosaggio Hb, ferritina, PCR. Valutazione audiologica. Visita ortopedica. Visita odontoiatrica. |
|
| Ogni 3 anni | Valutazione oculistica. | |
| In età adulta |
Come sopra, in più screening dell'osteoporosi:
|
|
| Ogni anno | Vaccino anti-influenzale. Screening della demenza a partire dai 40 anni. |
|
TERAPIA
Attualmente, non esiste alcun trattamento in grado di risolvere radicalmente la condizione di base. Tuttavia, è estremamente importante lavorare per il raggiungimento del maggior livello di qualità di vita possibile, grazie ai controlli medici che permettono di sorvegliare ed evidenziare le problematiche più comuni e favorirne l'inserimento nel contesto sociale e lavorativo. L'affettività e lo sviluppo interattivo dipendono fortemente dall'ambiente familiare e sociale. Il quoziente sociale può essere migliorato con tecniche di intervento precoce, sebbene i livelli perseguibili siano estremamente variabili. I bambini con SD dal punto di vista sociale mostrano competenze maggiori di quanto prevedibile in base al loro assetto cognitivo.
BIBLIOGRAFIA
- Devenny DA, et al. Normal ageing in adults with Down's syndrome: a longitudinal study. J Intellect Disabil Res 1996, 40: 208-21.
- Rubin SS, et al. Overweight prevalence in persons with Down syndrome. Ment Retard 1998, 36: 175-81.
- Percy ME, et al. Autoimmune thyroiditis associated with mild "subclinical" hypothyroidism in adults with Down syndrome: a comparison of patients with and without manifestations of Alzheimer disease. Am J Med Genet 1990, 36: 148-54.
- Capone G. Down syndrome. Current Management in Child Neurology 1999, 191-5.
- Chicoine B, et al. Development of a clinic for adults with Down syndrome. Ment Retard 1994, 32: 100-6.
- Brugge KL, et al. Cognitive impairment in adults with Down syndrome. Similarities to early cognitive changes in Alzheimer's disease. Neurology 1994, 44: 232-8.
- Burt DB, et al. Dementia in adults with Down syndrome: diagnostic challenges. Am J Ment Retard 1998, 103: 130-45.
- Cohen WI. Atlanto-axial instability. What's next? Arch Pediatr Adolesc Med 1998, 15: 119-22.
- Pilcher ES. Dental care for the patient with Down syndrome. Down Syndrome Research and Practice 1998, 5: 111-6.
- Smith D. Health care management of adults with Down syndrome. Am Fam Physician 2001, 15: 1031-8.
- Van Cleve SN, et al. Part II: clinical practice guidelines for adolescents and young adults with Down syndrome: 12 to 21 years. J Pediatr Health Care 2006, 20: 198-205.
Sindrome di Marfan
Angela Ida Pincelli
Clinica Medica, Università Milano-Bicocca, Ospedale San Gerardo, Monza
Epidemiologia e genetica
La sindrome di Marfan è un disordine del connettivo con interessamento multi-sistemico, a trasmissione autosomica dominante, con prevalenza di 2:10.000 e incidenza di 1 caso ogni 5000 individui (1). Il fenotipo peculiare che la caratterizza fu descritto per la prima volta nel 1896 dal pediatra francese Bernard Jean Antoine Marfan (2) in una giovane paziente di cinque anni, che presentava aracno-dattilia, dolico-stenomelia e cifo-scoliosi.
Solo nel 1991 venne identificata la causa molecolare, una mutazione del gene FBN1 a livello del cromosoma 15, codificante la fibrillina-1. Questa è la principale componente glicoproteica delle microfibrille nelle fibre elastiche, cruciale nel determinare l’elasticità dei tessuti e nel regolare la biodisponibilità del TGFβ, soprattutto nelle pareti dei grandi vasi, nei legamenti sospensori del cristallino, nelle vie aeree, nei legamenti para-vertebrali. L’alterazione strutturale delle microfibrille, insieme a una disregolazione dell’omeostasi della matrice extra-cellulare per eccessiva attività del TGFβ, determina un indebolimento del tessuto connettivo, alla base delle principali manifestazioni cliniche descritte nella sindrome (1,3). Mentre la maggior parte dei pazienti ha un familiare affetto, il 25% circa dei probandi presenta la malattia in forma sporadica, come risultato di una mutazione de novo.
Non é nota un’evidente correlazione fenotipo-genotipo: pazienti con la medesima mutazione spesso presentano un fenotipo variabile, tuttavia mutazioni a carico della porzione centrale del gene FBN1, in particolare il tratto compreso tra gli esoni 24-32, determinano una maggiore severità clinica (1,3).
Manifestazione cliniche
La graduale frammentazione delle lamelle elastiche, l’accumulo di muco-polisaccaridi e la riduzione delle cellule muscolari lisce nella parete dei grandi vasi (1,3-5) diminuiscono la distensibilità dell’aorta. Questa presenta maggiore fragilità e un rischio elevato di sviluppare una dilatazione aneurismatica, una delle manifestazioni cliniche di maggiore riscontro nella sindrome di Marfan, gravata da elevata morbilità per la possibile evoluzione verso la dissecazione e l’insufficienza della valvola aortica. La dilatazione, che interessa inizialmente la radice e il tratto prossimale dell’aorta ascendente, presenta un’eterogenea velocità di progressione, dalle forme neonatali rapidamente evolutive, ai casi più sfumati diagnosticati in età adulta. Anche i segmenti più distali possono andare incontro a dilatazione. Ampiamente descritta è la comparsa di dissecazione dell’aorta toracica discendente, soprattutto in pazienti precedentemente sottoposti a intervento chirurgico riparativo dell’aorta prossimale. Inoltre l’aneurisma dell’aorta addominale, manifestazione relativamente comune nella popolazione generale in età avanzata e in presenza di fattori scatenanti come aterosclerosi e fumo, presenta un maggiore rischio di dissecazione precoce nei soggetti con sindrome di Marfan.
Di frequente riscontro è il deterioramento strutturale dell’apparato valvolare, con la comparsa di prolasso della valvola mitrale, associato in alcuni soggetti a insufficienza valvolare, che, seppur raramente, può raggiungere un grado severo, fino a determinare un sovraccarico del ventricolo sinistro con evoluzione in cardiomiopatia dilatativa (3).
Il rischio di aritmie ventricolari e sopra-ventricolari, descritto in oltre il 10% dei pazienti con sindrome di Marfan, è un altro dato da non sottovalutare, in quanto il riscontro di aritmie ventricolari all’ECG dinamico rappresenta un forte valore predittivo di morte improvvisa (3).
A livello oculare la lussazione-sublussazione del cristallino (ectopia lentis) è presente in oltre il 60% dei pazienti. È spesso bilaterale, a esordio precoce nelle prime due decadi di vita, con una variabile espressione clinica: da forme asintomatiche a manifestazioni più severe, gravate da possibili complicanze, come glaucoma, cataratta precoce, uveite. Una diagnosi tempestiva è di fondamentale importanza, poiché una correzione tardiva è associata a scarso recupero dell’acuità visiva. Frequente è l’insorgenza di miopia. Manifestazione più rara, ma caratterizzata da elevata morbilità, è rappresentata dal distacco di retina (1,3).
Le anomalie muscolo-scheletriche, spesso evidenti in età pediatrica, sono i primi segni che più frequentemente portano il paziente all’attenzione del medico con il sospetto di sindrome di Marfan. In particolare, l’eccessiva crescita lineare, già presente nel corso della vita prenatale, determina una lunghezza alla nascita prossima al 90° percentile della popolazione generale. Nei primi tre anni di vita la crescita media si colloca tra il 50° e il 90° percentile, fino a superare successivamente il 95° percentile, con aspetto disarmonico per perdita delle proporzioni corporee: netta prevalenza degli arti sul tronco (dolico-stenomelia), rapporto tra il segmento superiore e il segmento inferiore di 1.05.
L’accelerazione di crescita puberale risulta precoce, raggiungendo il picco in anticipo, mediamente di circa 2.4 anni nei maschi e di 2.2 anni nelle femmine, che spesso presentano il menarca prima dei 12 anni. La statura finale supera il proprio bersaglio genetico e si attesta in media a 191 cm per maschi e 173 cm per le donne. L’età ossea è generalmente in linea con l’età cronologica in assenza di alterazioni degli assi ipotalamo-ipofisari (6).
Altre stigmate muscolo-scheletriche, polmonari, neurologiche e cutanee caratterizzano il fenotipo marfanoide, come descritto in tabella 1.
| Tabella 1 Criteri diagnostici per s. di Marfan |
||
| Criteri maggiori | Criteri minori | |
| Sistema cardiovascolare | Aneurisma aorta ascendente***/dissecazione | Prolasso mitralico Dilatazione arteria polmonare se età < 40 anni Calcificazione anello mitralico se età < 40 anni Dilatazione/dissecazione aorta toracica o addominale se età < 50 anni |
| Sistema oculare | Ectopia lentis | Cornea piatta Ipoplasia dei muscoli ciliari |
| Sistema nervoso | Dilatazione del canale lombo-sacrale (ectasia durale)*** | |
| Sistema muscolo-scheletrico (almeno 4 manifestazioni) | Pectus excavatum Pectus carenatum Rapporto segmento corporeo superiore/inferiore < 0.86* oppure apertura braccia/altezza > 1.05 Segno polso-pollice** Scoliosi > 20° o spondilo-listesi Ridotta estensione del gomito < 170° Dislocazione del malleolo mediale/piede piatto Protrusione acetabolare |
Pectus excavatum non severo (senza indicazione chirurgica) Iperlassità legamentosa Palato ogivale o affollamento dentario Dismorfismo facciale: dolico-cefalia, ipoplasia malare, enoftalmo, retrognazia, rima palpebrale obliqua lateralmente verso il basso |
| Familiarità/genetica | Familiare di I grado con s. di Marfan Mutazione FBN1 |
|
| Polmone | Enfisema apicale Pneumo-torace spontaneo |
|
| Cute | Strie atrofiche non correlate a variazioni ponderali o esiti di gravidanza Ernie recidivanti |
|
|
* Valori patologici del rapporto tra segmento corporeo superiore e segmento inferiore in età pediatrica:
** Il segno del pollice è positivo quando l’intera falange distale del pollice in posizione di adduzione si estende oltre il margine ulnare del palmo della mano. Il segno del polso è positivo quando la punta del pollice si sovrappone all’intera falange distale del quinto dito nel circondare il polso contro-laterale *** Diametro aortico, misurato a livello dei seni di Valsalva (generalmente mediante ecocardiogramma trans-toracico e in casi selezionati, con ecocardiogramma trans-esofageo, angioTC e RMN) > 2 deviazioni standard (Z-score) rispetto al valore medio descritto nella popolazione generale, utilizzando nomogrammi per età e superficie corporea |
||
In particolare, il pectus excavatum e la cifoscoliosi sono descritti in circa il 65% dei pazienti e nelle forme severe, possono determinare un deficit ventilatorio restrittivo. La dilatazione del canale lombo-sacrale (ectasia durale), generalmente asintomatica, può essere associata a lombalgia, cefalea da ipotensione endocranica e raramente a una disfunzione neurologica (incontinenza). Infine i pazienti con sindrome di Marfan hanno un alto rischio di sviluppare osteoporosi (1,3,7).
Diagnosi e follow-up
I criteri di Ghent, pubblicati nel 1996, hanno rappresentato per anni il gold-standard per la diagnosi di sindrome di Marfan, che in assenza di una familiarità positiva, veniva definita dalla presenza di un criterio maggiore in due apparati diversi (cardio-vascolare, scheletrico e oculare, includendo la mutazione di FBN1 come criterio maggiore), associato a un criterio minore, mentre in presenza di familiarità (familiare di I grado con diagnosi clinica), era necessario il riscontro di un solo criterio maggiore come descritto nella tabella 1. Tuttavia, la necessità di disporre di un approccio diagnostico più specifico ha portato a una revisione di tali criteri, pubblicata nel 2010, focalizzando l’attenzione sulla valutazione molecolare e sulla presenza delle alterazioni cardio-vascolari e oculari, mentre alle altre manifestazioni viene attribuito un punteggio diverso correlato al grado di specificità, inserito in uno score sistemico (tabella 2), che è considerato positivo se ≥ 7 (figura 1a e 1b) (5).
| Tabella 2 Calcolo dello score diagnostico sistemico |
||
| Caratteristica | Punteggio assegnato | Variazione punteggio |
| Segno del polso e del pollice di Steinberg | 3 | 1 se solo uno dei due segni |
| Pectus carenatum | 2 | 1 se pectus excavatum (meno specifico) |
| Deformità piede: dislocazione malleolo mediale | 2 | 1 se solo piede piatto |
| Pneumo-torace | 2 | |
| Ectasia durale | 2 | |
| Protrusione dell’acetabolo | 2 | |
| Ridotto rapporto segmento corporeo superiore/inferiore + incremento rapporto apertura braccia/altezza > 1.05 in assenza di severa scoliosi | 1 | |
| Scoliosi o cifosi toraco-lombare | 1 | |
| Ridotta estensione del gomito < 170° | 1 | |
| Dismorfismi facciali (3 su 5): dolico-cefalia, enoftalmo, micrognazia, ipoplasia malare, rima palpebrale obliqua lateralmente verso il basso | 1 | |
| Strie cutanee di atrofia | 1 | |
| Miopia > 3 diottrie | 1 | |
| Prolasso mitrale | 1 | |

Figura 1a. Nuovi criteri diagnostici in assenza di storia familiare positiva per S di Marfan

Figura 1b. Nuovi criteri diagnostici in presenza di storia familiare positiva per S di Marfan
* In assenza di una mutazione di FBN1 associata a sindrome di Marfan, la diagnosi è confermata previa esclusione di sindromi con possibile fenotipo marfanoide: sindrome di Shprintzen-Golberg, sindrome di Loeys-Dietz, variante vascolare della sindrome di Ehlers-Danlos e dopo valutazione molecolare dei geni TGFBR1, TGFB2, COL3A1 se necessario
** L’esecuzione di Rx bacino e RM pelvica in assenza di sintomatologia clinica è indicata solamente nel caso in cui il riscontro di protrusione acetabolare e di ectasia durale risulta necessario per soddisfare pienamente i criteri di Ghent e confermare la diagnosi di sindrome di Marfan
Tuttavia i criteri di Ghent non permettono di escludere la diagnosi di sindrome di Marfan in età pediatrica, a causa della penetranza età-correlata di molte manifestazioni fenotipiche. Pertanto, i pazienti che non soddisfano pienamente i criteri di Ghent necessitano di un adeguato follow-up, almeno fino al raggiungimento dell’età adulta. Inoltre, molte condizioni cliniche possono presentare un habitus marfanoide, a volte di difficile diagnosi differenziale (figura 2).

Figura 2. Diagnosi differenziale della sindrome di Marfan
Il risultato della valutazione molecolare deve sempre essere inquadrato nel contesto clinico, in quanto mutazioni a carico di FBN1 sono state descritte anche in altre fibrillinopatie con una prognosi migliore, come ectopia lentis familiare, aneurisma aortico toracico familiare, sindrome MASS (coinvolgimento Mitrale, Aorta, Scheletro e Skin); inoltre, l’assenza di mutazioni di FBN1 non esclude l’ipotesi di sindrome di Marfan, che nel 10% dei casi è secondaria a mutazione di TGFBR1 e TGFBR2, geni del recettore del TGFβ (5).
La diagnosi di sindrome di Marfan deve essere seguita da un costante follow-up multi-disciplinare sin dall’età evolutiva. Di cruciale importanza è la valutazione cardiologica per la diagnosi e il monitoraggio dell’aneurisma aortico (figura 3) (5).

Figura 3. Follow-up cardiologico della sindrome di Marfan
Inoltre, durante le visite pediatriche devono essere regolarmente controllati i parametri auxologici, utilizzando curve di crescita specifiche per sindrome di Marfan, in quanto velocità di crescita e timing dello spurt puberale si discostano dalle medie della popolazione generale, come descritto in precedenza. Uno stretto follow-up ortopedico deve accompagnare tutta l’età evolutiva, soprattutto nel corso dello sviluppo puberale, per elevato rischio di sviluppare cifosi, scoliosi e alterazioni toraciche a causa di un’eccessiva crescita costale. Con frequenza annuale va programmata una valutazione oculistica, mentre il controllo periodico del metabolismo calcio-fosforo è necessario soprattutto in presenza di altri fattori di rischio per osteoporosi.
Terapia
Grazie alle nuove potenzialità terapeutiche e al miglioramento delle metodiche diagnostiche, la sopravvivenza media dei pazienti affetti da sindrome di Marfan è attualmente quasi sovrapponibile alla popolazione generale e generalmente > 70 anni.
Il gold-standard della terapia medica è rappresentata dall’uso del ß-bloccante, il quale, grazie all’effetto inotropo e cronotropo negativo, riduce lo stress emodinamico sulla parete arteriosa e rallenta la progressione della dilatazione aortica (3-5). Il farmaco maggiormente utilizzato è l’atenololo, generalmente ben tollerato grazie alla lunga emivita e alla cardio-selettività. Nei pazienti che non tollerano il ß-bloccante, la terapia di seconda scelta è rappresentata dai calcio-antagonisti non-diidropiridinici (verapamil).
La terapia va iniziata in profilassi primaria in tutti pazienti che giungono all’osservazione per sindrome di Marfan con aneurisma della radice aortica e in profilassi secondaria in tutti i soggetti precedentemente sottoposti a intervento chirurgico correttivo al fine di proteggere le grandi arterie native. In età pediatrica la terapia risulta controversa nel caso di una dilatazione aortica con z-score compreso tra 2 e 2.5, visto il ridotto rischio di dissecazione nel bambino. Anche la terapia con losartan ha mostrato un impatto positivo sulla velocità di progressione dell’aneurisma, grazie all’inibizione del recettore I dell’angiotensina II, che si associa a una riduzione del segnale TGF-ß (8). Tuttavia, l’uso di sartani/ACE inibitori nella sindrome di Marfan è considerato al momento off-label in assenza di una cardiopatia strutturale o di ipertensione arteriosa.
In caso di insuccesso della terapia medica e ulteriore progressione dell’aneurisma, un ruolo cruciale è svolto dalla correzione chirurgica in elezione, che presenta un miglior esito clinico rispetto a un intervento in urgenza per dissecazione. Il timing del trattamento chirurgico profilattico è controverso e va individualizzato sulla base dei fattori di rischio per dissecazione, come dimensione aortica al seno di Valsalva > 5 cm, velocità di crescita > 0.5 cm/anno, familiarità per dissecazione, rapporto del diametro della radice aortica e del segmento discendente > 2 (3-5).
Contrariamente alla terapia delle complicanze cardio-vascolari, il trattamento di riduzione dell’eccessiva crescita, manifestazione di grande impatto prognostico quoad valetudinem, non ha mostrato risultati incoraggianti (9). In particolare, la terapia ormonale con steroidi sessuali ad alte dosi, somministrata dall’inizio dello sviluppo puberale fino alla chiusura delle cartilagini epifisarie, risulta caratterizzata da uno scarso profilo di sicurezza a lungo termine e associata a ridotta fertilità in età adulta (9,10).
Bibliografia
- Judge DP, Dietz HC. Marfan's syndrome. Lancet 2005, 366: 1965-76.
- Marfan AB. Un cas des deformation congenitale des quarte members plus prononcée aux extremités caracterisée par l’allongement des os avec un certin degrée d’amincissement. Bull Mem Soc Med Hop Paris 1896, 13: 220-6.
- Cook JR, Ramirez F. Clinical, diagnostic, and therapeutic aspects of the Marfan syndrome. Adv Exp Med Biol 2014, 802: 77-94.
- Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation 2008, 117: 2802-13.
- Summers KM, West JA, Hattam A, et al. Recent developments in the diagnosis of Marfan syndrome and related disorders. Med J Aust 2012, 197: 494-7.
- Erkula G, Jones KB, Sponseller PD, et al. Growth and maturation in Marfan syndrome. Am J Med Genet 2002, 109: 100-15.
- Grover M, Brunetti-Pierri N, Belmont J, et al. Assessment of bone mineral status in children with Marfan syndrome. Am J Med Genet A 2012, 158A: 2221-4.
- Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 2013, 34: 3491-500.
- Rozendaal L, le Cessie S, Wit JM, et al. Growth-reductive therapy in children with Marfan syndrome. J Pediatr 2005, 147: 674-9.
- Venn A, Bruinsma F, Werther G, et al. Oestrogen treatment to reduce the adult height of tall girls: long-term effects on fertility. Lancet 2004, 364: 1513-8.
Sindrome di Noonan
Antonino Crinò
SS Patologia Endocrina Autoimmune, UOC Endocrinologia e Diabetologia, Ospedale Bambino Gesù, IRCCS – Palidoro (Roma)
Epidemiologia e clinica
La sindrome di Noonan (SN) è una malattia genetica relativamente frequente, con incidenza stimata tra 1/1.000 e 1/2.500 nati (1). La sindrome può essere sporadica (circa il 60%), ma si può trasmettere anche come carattere autosomico dominante.
È caratterizzata da estrema variabilità clinica (2). Le manifestazioni cliniche più frequenti sono: bassa statura, facies dismorfica, difetti cardiaci congeniti (nel 70% circa dei pazienti) e altre comorbilità (anomalie a carico del sistema visivo, linfatico, coagulativo, genitale, scheletrico, ectodermico ed emopoietico) (tabella 1).
| Tabella 1 Caratteristiche cliniche della sindrome di Noonan |
|
| Anomalie fetali | Idrope fetale Idramnios/polidramnios |
| Addome | Ernia inguinale Epato-splenomegalia |
| Viso | Ipertelorismo con rime palpebrali rivolte verso il basso Micrognazia Epicanto Ptosi palpebrale Cheratocono Coloboma (iride, retina, macula) Collo corto Orecchie a basso impianto, retro-ruotate, con elice ispessito Palatoschisi, palato ogivale |
| Cardiopatie congenite | Stenosi della polmonare (50-60%) Stenosi aortica Coartazione aortica Ipoplasia cuore destro Prolasso-insufficienza mitralica Cardiomiopatia ipertrofica (RA1, BRAF)(10-20%) Difetti settali (6-10%) |
| Scheletro | Bassa statura Cubito valgo Fusione vertebrale/emivertebre Scoliosi (10-15%) |
| Cute e annessi | Lipo-atrofia/carenza tessuto sottocutaneo Lassità cute Edema/linfedema Unghie sottili ipoplasiche Bassa attaccatura capelli Anomalie dermatoglifi palmari |
| Anomalie genito-urinarie | Agenesia-ipoplasia renale Rene a ferro di cavallo Anomalie ureterali (reflusso/idronefrosi) Criptorchidismo Ipospadia/epispadia Micropene Ipogonadismo/pubertà ritardata |
| Sistema nervoso | Idrocefalo/dilatazione ventricoli cerebrali Malformazione di Arnold-Chiari Anomalie vascolari intra-craniche Ritardo mentale (borderline/lieve) Ipotonia Neuroblastoma |
| Anomalie toraciche | Petto carenato/escavato (70-95%) Capezzoli ipoplasici/assenti/sovrannumerari |
| Diatesi emorragica | Alterazioni delle piastrine/trombocitopenia con tendenza al sanguinamento Disordini della coagulazione |

Altri segni associati sono difficoltà di alimentazione durante l’infanzia e displasia linfatica. È riportata aumentata incidenza di leucemie (in particolare leucemia mielo-monocitica giovanile) o tumori solidi (3).
Genetica e diagnosi
In circa il 50% dei casi, la sindrome è causata da mutazioni del gene PTPN11 (12q24.1), che causano un guadagno di funzione della proteina tirosin-fosfatasi SHP-2, non recettoriale.
Recentemente, sono state identificate mutazioni in altri geni della via metabolica di RAS-MAPK (KRAS, SOS1, NRAS BRAF, RAF1, SHOC2 e CBL) in una piccola percentuale di pazienti con SN. I portatori della mutazione presentano maggiore incidenza di stenosi polmonare e diatesi emorragiche (4).
In circa il 75% dei casi è possibile confermare la diagnosi clinica con la ricerca di mutazioni in geni che codificano per alcune proteine della cascata RAS-MAPK.
La diagnosi avviene in base alle caratteristiche cliniche ed eventualmente all’analisi della storia familiare e può essere confermata con l’indagine genetica (tab 2). Il cariotipo è sempre normale nella SN (5).
| Tabella 2 Criteri per la diagnosi clinica della sindrome di Noonan |
||
| Caratteristiche | Maggiori | Minori |
| Faccia | Faccia dismorfica tipica | Faccia dismorfica suggestiva |
| Cuore | Stenosi della valvola polmonare Cardiomiopatia ostruttiva ipertrofica e/o tipiche alterazioni ECGrafiche |
Altri difetti |
| Statura | < 3° centile | < 10° centile |
| Torace | Pectus carenatum/excavatum | Torace ampio |
| Anamnesi familiare | Parente I grado di soggetto con diagnosi accertata di SN | Parente I grado di soggetto con diagnosi suggestiva di SN |
| Altro | Coesistenza dei seguenti 3 sintomi: criptorchidismo, ritardo mentale, displasia vasi linfatici | Uno dei seguenti 3 sintomi: criptorchidismo, ritardo mentale, displasia vasi linfatici |
Diagnosi accertata (5):
- faccia dismorfica tipica + 1 altro sintomo maggiore o + 2 sintomi minori;
- oppure faccia dismorfica suggestiva + 2 sintomi maggiori o + 3 altri sintomi minori.
La bassa statura è un carattere fenotipico più costante e caratteristico della SN, manifestandosi già in età pediatrica. Anche se la statura definitiva è al di sotto dei valori di normalità per la popolazione generale (in media 152.7 cm nelle femmine e 162.5 cm nei maschi), generalmente non si riscontra un deficit di secrezione dell'ormone della crescita pur in presenza di bassi livelli di IGF-1 (6). Si suppone che una resistenza periferica al GH (di tipo post-recettoriale) potrebbe essere conseguenza della mutazione (in particolare di PTPN11) e giustificare la scarsa risposta al trattamento con GH riscontrata nella maggior parte dei pazienti. Probabilmente mutazioni diverse a carico di tale gene potrebbero giustificare sia la diversa gravità di iposomia che la diversa risposta al trattamento con GH (7). La cardiomiopatia ipertrofica rappresenta una controindicazione assoluta alla terapia con GH. Lo sviluppo puberale di questi pazienti è quasi sempre ritardato (8).
La diagnosi differenziale si pone con la sindrome di Turner, la sindrome cardio-facio-cutanea, la sindrome di Costello, la neurofibromatosi tipo 1 (NF1) e la sindrome di LEOPARD. Tutte queste patologie vengono incluse oggi nel capitolo più vasto della rasopatie (9).
Terapia
I pazienti affetti da SN richiedono un attento follow-up multidisciplinare periodico, per poter garantire una diagnosi precoce di interessamento d'organo, prevenire le complicanze, migliorare la prognosi e offrire le opportunità terapeutiche adeguate.
Gli obiettivi della presa in carico e del trattamento sono:
- garantire un attento follow-up multi-specialistico e individualizzato, per migliorarne la gestione e la storia naturale;
- fornire un adeguato trattamento farmacologico o chirurgico per le complicanze d'organo;
- fornire un corretto supporto psicologico.
La gestione dovrebbe essere rivolta alle difficoltà dell’alimentazione nella prima infanzia, e comprendere una valutazione della funzione cardiaca, della crescita e dello sviluppo motorio. Sono sempre considerate opportune fisioterapia e logopedia. Durante i primi anni di scuola deve essere effettuato un esame completo dell’occhio e una valutazione uditiva (10).
Per quanto riguarda le possibilità di cura, bisogna considerare che alcune lesioni cardiache devono essere corrette per via chirurgica (deve essere sempre effettuato una studio pre-operatorio della coagulazione), mentre in alcuni casi selezionati è consigliata la somministrazione di ormone della crescita.
Con cure e consulenze specialistiche, la maggior parte dei bambini con SN ha crescita e funzioni normali. Segni e sintomi si attenuano con l’età e molti adulti con SN non richiedono cure mediche specialistiche nell’età adulta.
Bibliografia
- Bhambhani V, Muenke M. Noonan syndrome. Am Fam Physician 2014, 89: 37-43.
- Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 2010, 126: 746-59.
- Roberts AE, Allanson JE, Tartaglia M, et al. Noonan syndrome. Lancet 2013, 381: 333-42.
- Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 2011, 25: 161-79.
- van der Burgt I. Noonan syndrome. Orphanet J Rare Dis 2007, 2: 4.
- Otten BJ, Noordam C. Growth in Noonan syndrome. Horm Res 2009, 72 suppl 2: 31-5.
- Dahlgren J. GH therapy in Noonan syndrome: review of final height data. Horm Res 2009, 72 suppl 2: 46-8.
- Kelnar CJH. Noonan syndrome: the hypothalamo-adrenal and hypothalamo-gonadal axes. Horm Res 2009, 72 suppl 2: 24-30.
- Pierpont ME, Magoulas PL,Adi S, et al. Cardio-facio-cutaneous syndrome: clinical features, diagnosis, and management guidelines. Pediatrics 2014, 134: e1149-62.
- Chacko E, Graber E, Regelmann MO, et al. Update on Turner and Noonan syndromes. Endocrinol Metab Clin North Am 2012, 41: 713-34.
Sindrome di Prader-Willi
Antonino Crinò
SS Patologia Endocrina Autoimmune, UOC Endocrinologia e Diabetologia, Ospedale Bambino Gesù, IRCCS – Palidoro (Roma)
Epidemiologia e genetica
La sindrome di Prader-Willi (PWS) è una patologia multi-sistemica congenita, che colpisce in eguale misura entrambi i sessi, con una prevalenza nella popolazione generale di circa 1:25.000. La sua reale prevalenza è comunque sottostimata a causa dell’ancora scarsa conoscenza della sindrome.
Rappresenta la forma più comune di obesità genetica e costituisce un esempio paradigmatico di malattia da difetto dell’imprinting (1). È causata da un’alterazione del cromosoma 15 di origine paterna (delezione: 70% dei casi; disomia uniparentale materna-UPD: circa 28% dei casi; difetto dell’imprinting center: circa l’1-2%). In rari casi la sindrome è dovuta a traslocazioni o riarrangiamenti cromosomici.
Clinica
Il quadro clinico completo non è presente fin dalla nascita; è infatti estremamente variabile a seconda dell’età in cui viene visto il paziente e alcuni dei segni clinici più caratteristici si renderanno più evidenti negli anni successivi (2).
In epoca fetale si osserva una riduzione dei movimenti fetali, in parte secondaria a una grave ipotonia muscolare che caratterizza il periodo neonatale e la prima infanzia, determinando, tra l’altro, difficoltà di suzione (spesso alimentazione con gavage), pianto flebile, scarso accrescimento ponderale e ritardo delle principali tappe dello sviluppo psico-motorio (3). Nel maschio, è quasi costante il criptorchidismo. Già in questa fase è possibile rilevare le tipiche dismorfie, tra cui dolico-cefalia, fronte prominente con diametro bifrontale stretto, occhi a mandorla, labbro superiore sottile e angoli della bocca rivolti in basso (4).

Figura 1: evidente ipotonia

Figura 2: evidenti le caratteristiche dismorfiche del volto
Verso i 2-3 anni si assiste, accanto a un miglioramento dell'ipotonia, alla comparsa di iperfagia ingravescente, secondaria a mancanza del senso di sazietà, che determina l'instaurarsi di obesità di grado anche molto elevato, particolarmente resistente al trattamento dietetico e farmacologico (5). L'obesità viene favorita inoltre dal basso dispendio energetico dovuto alla riduzione della massa magra. È in questa seconda fase che la malattia comincia ad essere più manifesta, con quella costellazione di disturbi che la caratterizzano: bassa statura, scoliosi, apnee notturne e narcolessia (soprattutto nei pazienti particolarmente obesi), ritardo cognitivo e del linguaggio (di varia entità), ipogonadismo e ritardo puberale (6), alterazioni neuro-comportamentali, tratti somatici caratteristici. Altri segni clinici sono acromicria (mani e/o piedi piccoli), saliva densa e vischiosa associata a importante carie dentaria, instabilità della temperatura corporea, elevata soglia del dolore, strabismo e lipodistrofia agli arti inferiori (4). Un'altra peculiare abitudine di questi bambini è quella di "stuzzicare" di continuo ogni minima escoriazione della pelle, determinando così la formazione di lesioni cutanee croniche con rischio di infezione (skin-picking) (5).

Figura 3: tipiche lesioni cutanee
In tali pazienti è presente una disregolazione dell'asse ipotalamo-ipofisi, che è causa di alterazioni della secrezione dell'ormone della crescita (GH), della funzione surrenalica, tiroidea, gonadica, dell'iperfagia e dell'obesità (7).
L’età adolescenziale rappresenta la fase critica della malattia, in cui si fanno più evidenti i tipici disturbi comportamentali: il paziente con PWS cerca il cibo in maniera ossessiva e ha frequenti scatti d’ira che possono rendere difficile, se non impossibile, la vita in famiglia e condizionare negativamente le relazioni sociali (8).

Figura 4: lipodistrofia arti inferiori

Figura 5
Completa questo quadro clinico, estremamente complesso, la presenza di complicanze metaboliche (diabete mellito di tipo 2, iperlipemia, ecc) e cardio-respiratorie, più frequenti nei soggetti con obesità severa (9). Molto importanti le complicanze osteo-articolari (scoliosi, cifosi, ecc) (10).
Dal punto di vista endocrino, è pressoché costante il rilievo di una ridotta crescita staturale, riconducibile nella maggior parte dei casi a una disfunzione dell’asse GH/IGF-I, accanto alla presenza di ipogonadismo e pubertà ritardata o incompleta (6), con conseguente sviluppo precoce di osteoporosi (11,12). In tali pazienti è stato inoltre riscontrato con frequenza variabile iposurrenalismo di origine centrale (13).
Nel soggetto adulto la prognosi può peggiorare ulteriormente a causa dell’accentuazione delle problematiche comportamentali (disturbi ossessivo-compulsivi, atteggiamenti rigidi, insistenti e irragionevoli, tendenza alla menzogna e al furto, bruschi mutamenti d’umore, esplosioni d’ira con talora reazioni violente e atti di auto-lesionismo), già evidenti a partire dall’età scolare, che insieme al deficit cognitivo condizionano negativamente le relazioni sociali e la possibilità di condurre una vita indipendente (14).
Diagnosi
Di fronte a un bambino con ipotonia alla nascita bisogna escludere le altre possibili cause, ma è importante pensare sempre alla PWS (15). Il sospetto clinico deve essere confermato dalle indagini genetiche: il test di metilazione insieme al cariotipo sono in grado di confermare la diagnosi di PWS nel 99% dei casi. Il rischio di ricorrenza per la PWS in successive gravidanze di genitori con figli affetti è solitamente < 1% (16)(tab 1).
| Tabella 1 Indicazioni per l'invio alla diagnosi genetica (sospetto PWS) (modif da 2) |
|
| Età | Caratteristiche cliniche |
| 0-2 anni | Ipotonia generalizzata + suzione ipovalida, pianto flebile, criptorchidismo |
| 2-6 anni | Ipotonia + anamnesi positiva per suzione ipovalida, pianto flebile + ritardo globale di sviluppo neuro-motorio, criptorchidismo, bassa statura con progressivo incremento ponderale |
| 6-12 anni | Anamnesi di ipotonia (che spesso persiste) e suzione debole + ritardo globale di sviluppo psico-motorio + iperfagia e ossessività verso il cibo con obesità centrale (se non controllata) |
| > 13 anni (fino età adulta) | Deficit cognitivo (solitamente ritardo mentale di grado lieve-moderato) + iperfagia con obesità centrale (se non controllata) + ipogonadismo e ritardo puberale + disturbi comportamentali tipici (accessi d’ira e manifestazioni compulsive di vario tipo) + anamnesi positiva per ipotonia, suzione ipovalida, pianto flebile |
In tabella 2 e 3 il percorso clinico-assistenziale e le indagini strumentali da effettuare nei pazienti con PWS a seconda dell'età.
| Tabella 2 Percorso clinico-assistenziale nella PWS |
||||
| Valutazione | 0-3 anni | 3-10 anni | 10-18 anni | > 18 anni |
| Clinica e auxologica (statura, peso, BMI, PA, circonferenze, ecc) | ogni 3-4 mesi | ogni 6 mesi | ogni 6 mesi | ogni 12 mesi (ogni 6 mesi se GH e/o incremento di peso) |
|
Glicemia/insulinemia a digiuno - HbA1c |
prima di terapia con GH (ogni 6 mesi in terapia con GH) | ogni 6-12 mesi (in particolare se terapia con GH) | ogni 12 mesi (ogni 6 mesi se alterazioni e/o GH) | ogni 12 mesi (ogni 6 mesi se GH e/o incremento di peso) |
| Assetto lipidico, epatico, uricemia | annuale | ogni 6-12 mesi (in particolare se terapia con GH) | ogni 12 mesi (ogni 6 mesi se alterazioni e/o GH) | ogni 12 mesi (ogni 6 mesi se GH e/o incremento di peso) |
| Funzione tiroidea (FT4, TSH) | epoca neonatale, prima di iniziare terapia con GH (poi ogni 6 mesi se in terapia) |
ogni 12 mesi (ogni 6 mesi se terapia con GH) | ogni 12 mesi (ogni 6 mesi se terapia con GH) | ogni 12 mesi (ogni 6 mesi se GH e/o incremento di peso) |
| Metabolismo calcio/fosforo | annuale | ogni 1-2 anni (più spesso se osteopenia/terapia) | ogni 1-2 anni (più spesso se osteopenia/terapia) | ogni 1-2 anni (più spesso se osteopenia/terapia) |
| Test da carico orale glucosio (OGTT) | no | ogni 12 mesi (in particolare se obesità e/o terapia con GH) | ogni 12 mesi (se sovrappeso e/o GH) | ogni 12 mesi (ogni 6 mesi se GH e/o incremento di peso) |
| GnRH test, testosterone/ estradiolo | no | no | dopo i 12-13 anni (su indicazione clinica) | su base clinica |
| Test di stimolo per GH + IGF-I, IGF-BP3 | se possibile | una sola volta | no | retesting per eventuale terapia GH |
| ACTH test | se possibile | una sola volta | ogni 12-24 mesi | ogni 12-24 mesi |
| Tabella 3 Indagini strumentali nella PWS |
||||
| Valutazione | 0-3 anni | 3-10 anni | 10-18 anni | > 18 anni |
| Età ossea | una sola volta | ogni 12 mesi (se terapia con GH) | ogni 12-24 mesi (su indicazione endocrinologica) | no |
| Poli-sonnografia + monitoraggio CO2 | se possibile (e comunque prima della terapia con GH) | ogni 12 mesi (su indicazione pneumologica/ORL) | ogni 12 mesi (su indicazione pneumologica/ORL) | ogni 24 mesi (ogni 12 mesi se OSAS) |
| DEXA (composizione corporea) | no | ogni 12 mesi | ogni 24 mesi (se non alterazioni) | ogni 24 mesi (se osteopenia) o 48 mesi (se normale) |
| Rx colonna | ogni 12 mesi | ogni 12 mesi (in particolare se in terapia con GH) | ogni 12 mesi (su indicazione ortopedica) | su base clinica |
|
Ecografia addominale |
una sola volta | ogni 12 mesi (sulla base dei risultati degli esami) | ogni 24 mesi (se non alterazioni) | ogni 24 mesi (ogni 12 mesi se litiasi biliare/steatosi) |
Terapia
È fondamentale iniziare la terapia con GH al più presto possibile e comunque entro l'anno di vita, per gli effetti positivi che si ottengono sull’ipotonia, sulla composizione corporea, sullo sviluppo psico-motorio e sull’aspetto cognitivo, nonchè sul fenotipo. I benefici del trattamento con GH a piccole dosi nei pazienti adulti è stato più volte validato (17,18).
I pazienti riconosciuti precocemente e trattati fin dai primi mesi di vita con GH, nonchè avviati subito a un determinato percorso assistenziale e terapeutico multi-specialistico, oggi non sono obesi e presentano molto meno i tipici disturbi comportamentali (19).
Le problematiche respiratorie (apnee ostruttive e centrali, narcolessia, ecc) costituiscono un serio problema clinico che mette a rischio di morte improvvisa, in particolare i pazienti obesi, con ipertrofia adeno-tonsillare e marcata ipotonia.
La prevenzione dell'obesità deve pertanto essere iniziata precocemente fin dai primi anni di vita, in quanto successivamente qualsiasi trattamento è destinato a fallire. Negli adolescenti con obesità grave e ingravescente l'uso del palloncino intra-gastrico può determinare un controllo del peso corporeo, possibilmente in preparazione di un successivo intervento di chirurgia bariatrica (20).
La malattia è comunque quasi sempre invalidante e ha un decorso cronico ma con possibilità di miglioramento; alcuni pazienti devono essere assiduamente sorvegliati, specialmente quando rubano continuamente il cibo e presentano disturbi comportamentali più o meno gravi. In assenza di adeguato trattamento, i soggetti affetti sono destinati a vivere una vita di emarginazione e disagio psico-sociale e a sviluppare gravi complicanze, che li possono condurre a morte prematura.
È stato evidenziato come il trattamento precoce sia in grado di modificare la storia naturale della malattia e la prognosi (19). Date le differenti problematiche dei pazienti con PWS, si sottolinea pertanto la fondamentale importanza di coinvolgere diverse figure professionali e specialistiche fin dal periodo neonatale, al fine di prevenire l’insorgenza dell'obesità e delle sue complicanze, migliorando l’outcome e la qualità di vita del paziente (tab 4). È fondamentale, inoltre, creare una buona relazione e collaborazione tra medico e familiari, chiarendo che nessuno dei problemi associati alla PWS può essere gestito e risolto da un unico trattamento, ma che è necessario attuare un programma assistenziale e terapeutico multidisciplinare. La complessità del quadro clinico e la sua rarità richiedono inoltre l'identificazione di Centri di riferimento, allo scopo di creare dei team multidisciplinari di esperti e rendere in questo modo le decisioni diagnostiche e gli interventi terapeutici più coordinati ed efficaci.
| Tabella 4 Assistenza multi-specialistica al paziente PWS |
||||
| Valutazione | 0-3 anni | 3-10 anni | 10-18 anni | > 18 anni |
| Endocrinologo | ogni 6-12 mesi | ogni 6-12 mesi | ogni 6-12 mesi | ogni 12 mesi (6 mesi se GH) |
| Dietista/dietologo | ogni 6-12 mesi (più frequente se già sovrappeso) | ogni 6-12 mesi (più frequente se sovrappeso) | ogni 6-12 mesi (più frequente se sovrappeso) | ogni 6-12 mesi (più frequente se obesità) |
| Ortopedico | prima della dimissione dalla neonatologia, poi ogni 6-12 mesi | ogni 6-12 mesi (su base clinica) | ogni 6-12 mesi (su base clinica) | su base clinica |
| Psicologo | sì (alla diagnosi) | ogni 12 mesi | ogni 12 mesi (su base clinica) | in base alle problematiche |
| ORL | in presenza di apnee ostruttive (prima della terapia con GH) | ogni 12 mesi (se non OSAS) | ogni 12 mesi (se non OSAS) | ogni 12 mesi |
| Bronco-pneumologo | no | ogni 12 mesi (se non OSAS) | ogni 12 mesi (se non OSAS) | ogni 24 mesi (12 mesi se GH e/o OSAS) |
| Chirurgo | sì (se criptorchidismo | sì (se criptorchidismo | su indicazione clinica (chirurgia bariatrica) | su indicazione clinica (chirurgia bariatrica) |
| Cardiologo (ECG/ eco-cardiogramma) | 1 sola volta | variabile su base clinica | ogni 24 mesi (più frequente se sovrappeso e/o OSAS) | ogni 12-24 mesi (più frequente se sovrappeso e/o OSAS) |
| Oculista | ogni 6-8 mesi | se strabismo o altri problemi | ogni 12 mesi | ogni 12 mesi se diabete |
| Logopedista | no | su indicazione | su base clinica | no |
| Odontoiatra | dopo i 2 anni e poi ogni 12 mesi | ogni 6 mesi | ogni 6 mesi | ogni 6 mesi |
| Neuro-psichiatra | ogni 3-6 mesi | ogni 12 mesi (più frequente se disturbi del linguaggio) | ogni 12 mesi (su base clinica) | ogni 12-24 mesi (in base alle problematiche) |
| Dermatologo | no | se skin-picking | in base alle problematiche | in base al quadro clinico |
Bibliografia
- Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med 2012, 14: 10–26.
- Gunay-Aygun M, Schwartz S, Heeger S, et al. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics 2001, 108: E92.
- Tuysuz B, Kartal N, Erener-Ercan T, et al. Prevalence of Prader-Willi syndrome among infants with hypotonia. J Pediatr 2014, 164: 1064-7.
- Crinò A, Di Giorgio G, Livieri C, et al. A survey on Prader-Willi syndrome in the Italian population: prevalence of historical and clinical signs. J Pediatr Endocrinol Metab 2009, 22: 883-93.
- Grugni G, Crinò A, Bosio L, et al. The Italian National Survey for Prader-Willi syndrome: an epidemiologic study. Am J Med Genet A 2008, 146: 861–72.
- Radicioni AF, Di Giorgio G, Grugni G, et al. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader-Willi syndrome. Clin Endocrinol 2012, 76: 72-7.
- Emerick JE,Vogt KS. Endocrine manifestations and management of Prader-Willi syndrome. Int J Pediatr Endocrinol 2013, 2013: 14.
- Sinnema M, Maaskant MA, van Schrojenstein Lantman-de Valk HM, et al. Physical health problems in adults with Prader-Willi syndrome. Am J Med Genet A 2011, 155A: 2112-24.
- Grugni G, Crinò A, Bedogni G, et al. Metabolic syndrome in adult patients with Prader-Willi syndrome. Nutr Metab Cardiovasc Dis 2013, 23: 1134-40.
- Nakamura Y, Murakami N, Iida T, et al. The characteristics of scoliosis inPrader-Willi syndrome(PWS): analysis of 58 scoliosis patients with PWS. J Orthop Sci 2015, 20: 17-22.
- Grugni G, Marzullo P, Ragusa L, et al. Impairment of GH responsiveness to combined GH-releasing hormone and arginine administration in adult patients with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2006, 65: 492-9.
- Di Giorgio G, Grugni G, Fintini D, et al. Growth hormone response to standard provocative stimuli and combined tests in very young children with Prader-Willi syndrome. Horm Res Paediatr 2014, 81: 189-95.
- Grugni G, Beccaria L, Corrias A, et al. Central adrenal insufficiency in young adults with Prader-Willi syndrome. Clin Endocrinol (Oxf) 2013, 79: 371-8.
- Hauber M, Stratmann B, Hoedebeck-Stuntebeck N, et al. Medical management for adults with Prader-Willi syndrome. Metab Syndr Relat Disord 2013, 11: 392-6.
- Trifirò G, Livieri C, Bosio L, et al. Genetic Obesity Study Group of the Italian Society of Paediatric Endocrinology and Diabetology. Neonatal hypotonia: don't forget the Prader-Willisyndrome. Acta Paediatr 2003, 92: 1085-9.
- Goldstone AP, Holland AJ, Hauffa BP, et al. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab 2008, 93: 4183-97.
- Deal CL, Tony M, Höybye C, et al. Growth Hormone Research Society workshop summary: consensus guidelines for recombinant human growth hormone therapy in Prader-Willi syndrome. J Clin Endocrinol Metab 2013, 98: E1072-87.
- Bakker NE, Kuppens RJ, Siemensma EP, et al. Eight years of growth hormone treatment in children with Prader-Willi syndrome: maintaining the positive effects. J Clin Endocrinol Metab 2013, 98: 4013-22.
- Bacheré N, Diene G, Delagnes V, et al. Early diagnosis and multidisciplinary care reduce the hospitalization time and duration of tube feeding and prevent early obesity in PWS infants. Horm Res 2008, 69: 45-52.
- De Peppo F, Di Giorgio G, Germani M, et al. BioEnterics intragastric balloon for treatment of morbid obesity in Prader-Willi syndrome: specific risks and benefits. Obes Surg 2008, 18: 1443-9.